Zhuang Liwei, Wu Yun, Han Jiwu, Ling Xiaohua, Wang Liguo, Zhu Chengyan, Fu Yili
State Key Laboratory of Robotics and System, Bio-X Centre, Harbin Institute of Technology, Harbin, Heilongjiang 150001, China ; Department of Gastroenterology, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150001, China.
Department of Gastroenterology, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150001, China.
Biomed Res Int. 2014;2014:278956. doi: 10.1155/2014/278956. Epub 2014 May 14.
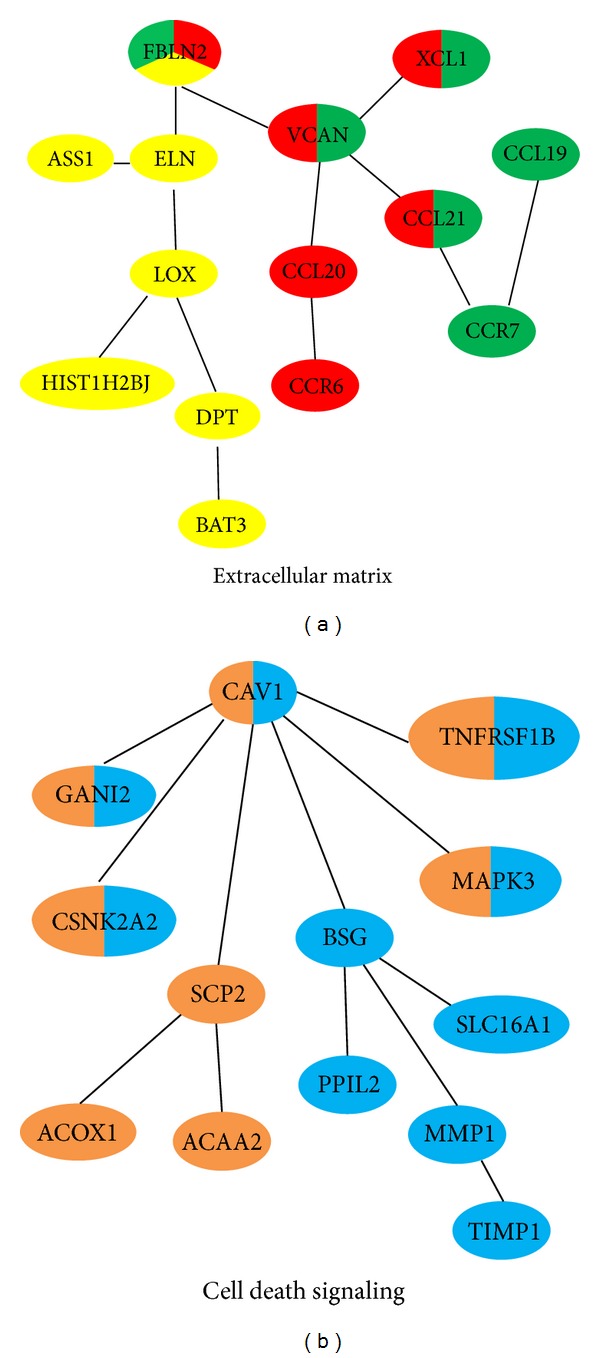
In recent years, high throughput technologies such as microarray platform have provided a new avenue for hepatocellular carcinoma (HCC) investigation. Traditionally, gene sets enrichment analysis of survival related genes is commonly used to reveal the underlying functional mechanisms. However, this approach usually produces too many candidate genes and cannot discover detailed signaling transduction cascades, which greatly limits their clinical application such as biomarker development. In this study, we have proposed a network biology approach to discover novel biomarkers from multidimensional omics data. This approach effectively combines clinical survival data with topological characteristics of human protein interaction networks and patients expression profiling data. It can produce novel network based biomarkers together with biological understanding of molecular mechanism. We have analyzed eighty HCC expression profiling arrays and identified that extracellular matrix and programmed cell death are the main themes related to HCC progression. Compared with traditional enrichment analysis, this approach can provide concrete and testable hypothesis on functional mechanism. Furthermore, the identified subnetworks can potentially be used as suitable targets for therapeutic intervention in HCC.
近年来,诸如微阵列平台等高通量技术为肝细胞癌(HCC)研究提供了一条新途径。传统上,常用生存相关基因的基因集富集分析来揭示潜在的功能机制。然而,这种方法通常会产生过多的候选基因,并且无法发现详细的信号转导级联反应,这极大地限制了它们在生物标志物开发等临床应用中的使用。在本研究中,我们提出了一种网络生物学方法,从多维组学数据中发现新的生物标志物。这种方法有效地将临床生存数据与人类蛋白质相互作用网络的拓扑特征以及患者表达谱数据结合起来。它可以产生基于网络的新生物标志物以及对分子机制的生物学理解。我们分析了80个HCC表达谱阵列,并确定细胞外基质和程序性细胞死亡是与HCC进展相关的主要主题。与传统的富集分析相比,这种方法可以提供关于功能机制的具体且可检验的假设。此外,所识别的子网有可能用作HCC治疗干预的合适靶点。