Shaw P J, Agard D A, Hiraoka Y, Sedat J W
Department of Cell Biology, John Innes Institute, Norwich, United Kingdom.
Biophys J. 1989 Jan;55(1):101-10. doi: 10.1016/S0006-3495(89)82783-3.
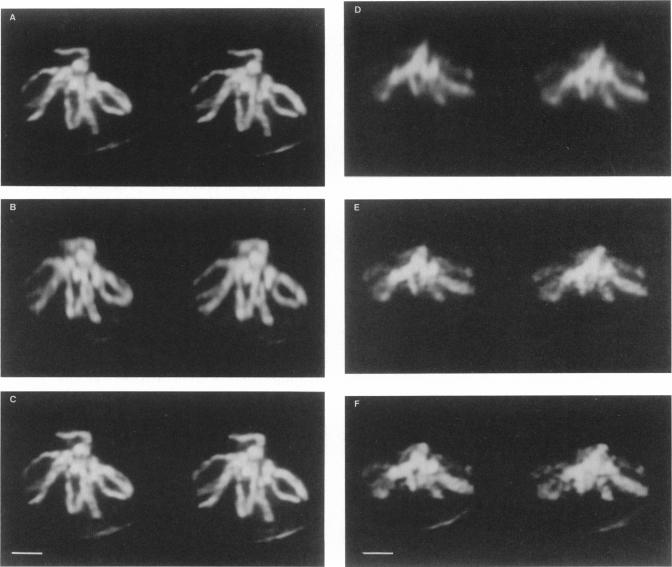
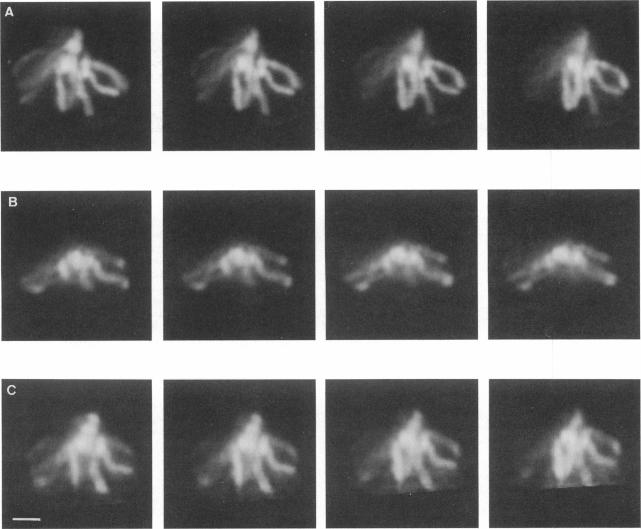
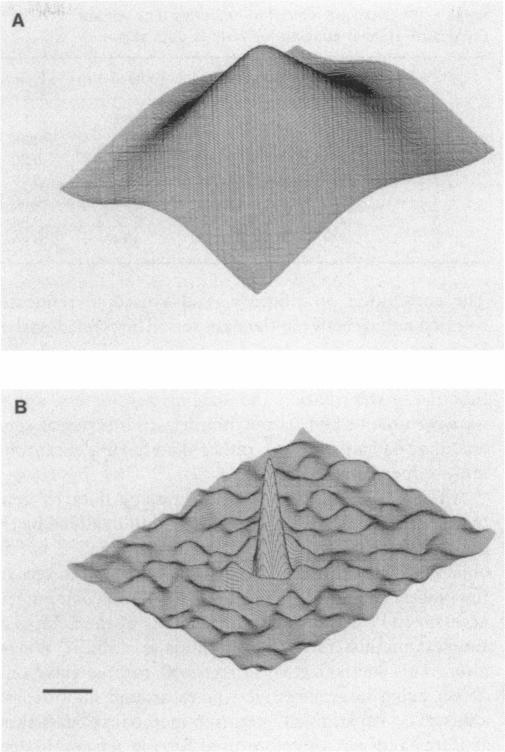
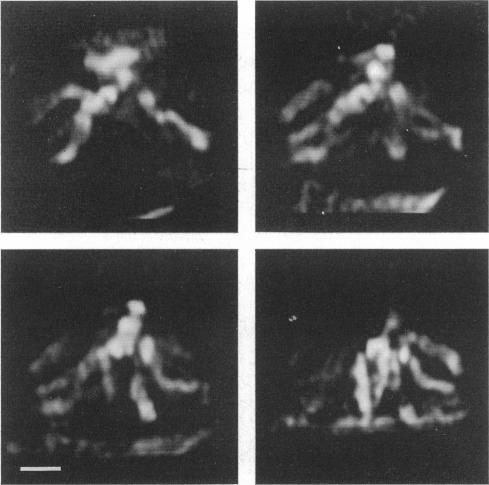
The resolution along the optical axis (z) is much less than the in-plane resolution in any current optical microscope, conventional or otherwise. We have used mutually tilted, through-focal section views of the same object to provide a solution to this problem. A tilting specimen stage was constructed for an optical microscope, which with the use of a coverslip-free water immersion lens, allowed the collection of data sets from intact Drosophila melanogaster embryos at viewing directions up to 90 degrees apart. We have devised an image processing scheme to determine the relative tilt, translation, and sampling parameters of the different data sets. This involves the use of a modified phase cross-correlation function, which produces a very sharp maximum. Finally the data sets are merged using figure-of-merit and local area scaling techniques borrowed from x-ray protein crystallography. We demonstrate the application of this technique to data sets from a metaphase plate in an embryo of Drosophila melanogaster. As expected, the merged reconstruction combined the highest resolution available in the individual data sets. As estimated from the Fourier transform, the final resolution is 0.25 microns in x and y and 0.4 microns in z. In the final reconstruction all ten chromosome arms can be easily delineated; this was not possible in the individual data sets. Within many of the arms the two individual chromatids can be seen. In some cases the chromatids are wrapped around each other helically, in others they lie alongside each other in a parallel arrangement.
在任何当前的光学显微镜中,无论是传统的还是其他类型的,沿光轴(z)方向的分辨率都远低于平面内分辨率。我们利用对同一物体相互倾斜的焦平面截面视图来解决这个问题。为光学显微镜构建了一个倾斜载物台,使用无盖玻片的水浸透镜,能够从完整的黑腹果蝇胚胎中以相差达90度的观察方向收集数据集。我们设计了一种图像处理方案来确定不同数据集的相对倾斜、平移和采样参数。这涉及使用一种改进的相位互相关函数,该函数会产生一个非常尖锐的最大值。最后,利用从X射线蛋白质晶体学借鉴的品质因数和局部区域缩放技术将数据集合并。我们展示了该技术在黑腹果蝇胚胎中期板数据集上的应用。正如预期的那样,合并后的重建结合了各个数据集中可用的最高分辨率。根据傅里叶变换估计,最终分辨率在x和y方向为0.25微米,在z方向为0.4微米。在最终重建中,可以轻松勾勒出所有十条染色体臂;这在单个数据集中是不可能的。在许多染色体臂内,可以看到两条单独的染色单体。在某些情况下,染色单体相互螺旋缠绕,在其他情况下,它们相互平行排列。