Lao Brooke Bullock, Drew Kevin, Guarracino Danielle A, Brewer Thomas F, Heindel Daniel W, Bonneau Richard, Arora Paramjit S
Department of Chemistry and ‡Departments of Biology and Computer Science, New York University , New York, New York 10003, United States.
J Am Chem Soc. 2014 Jun 4;136(22):7877-88. doi: 10.1021/ja502310r. Epub 2014 May 23.
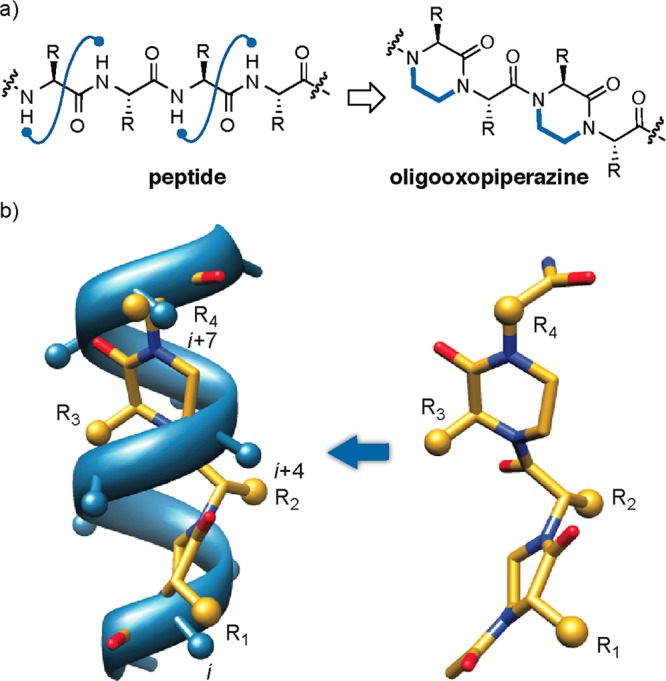
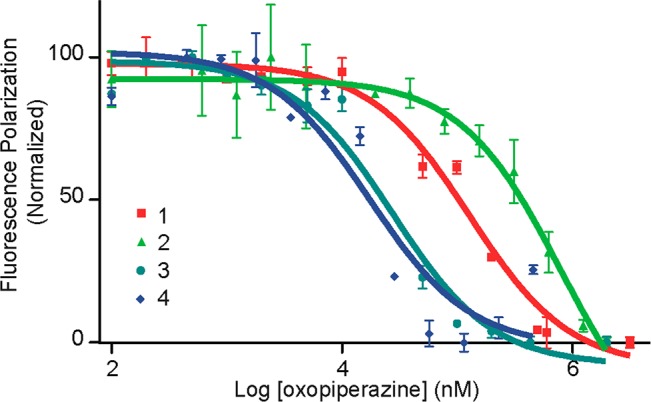
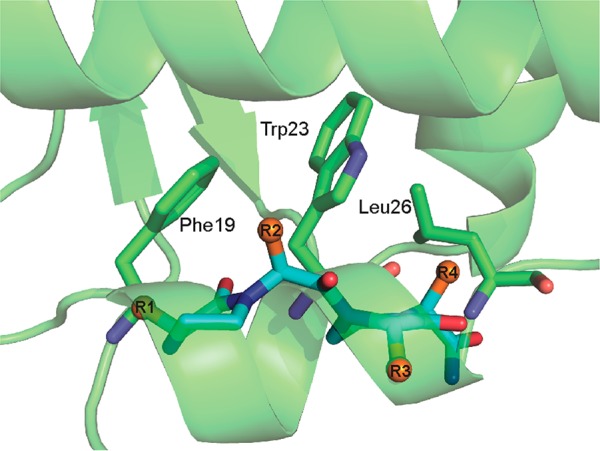
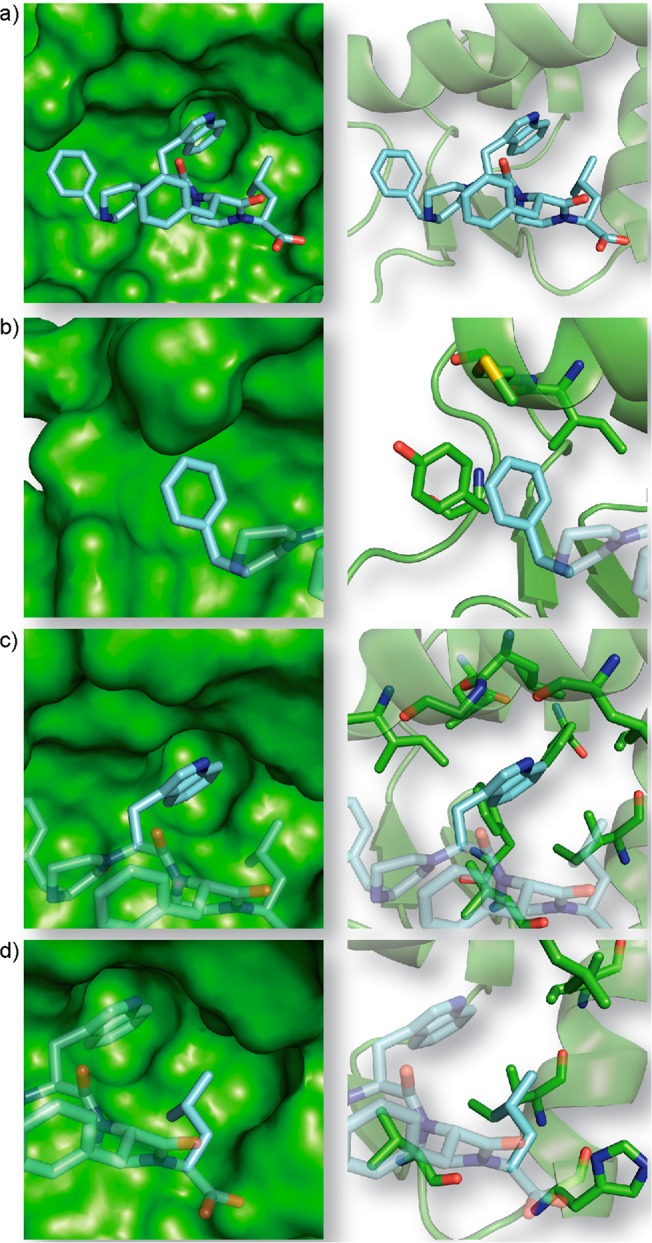
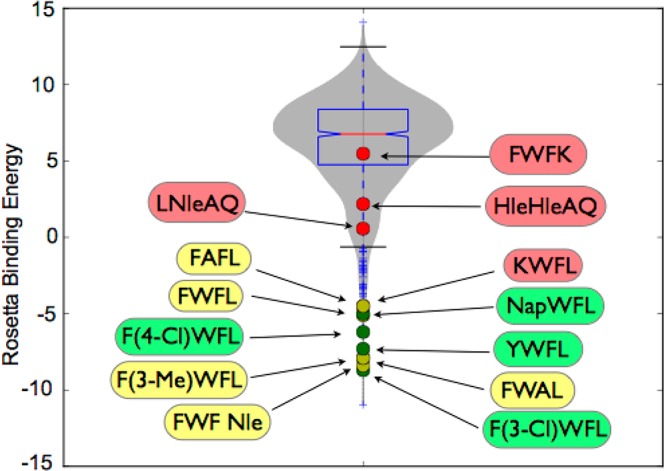
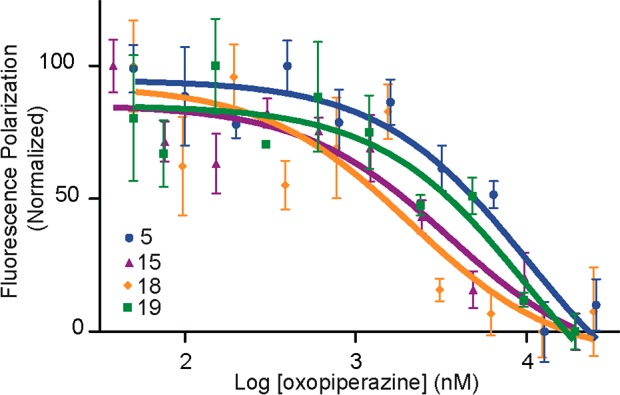
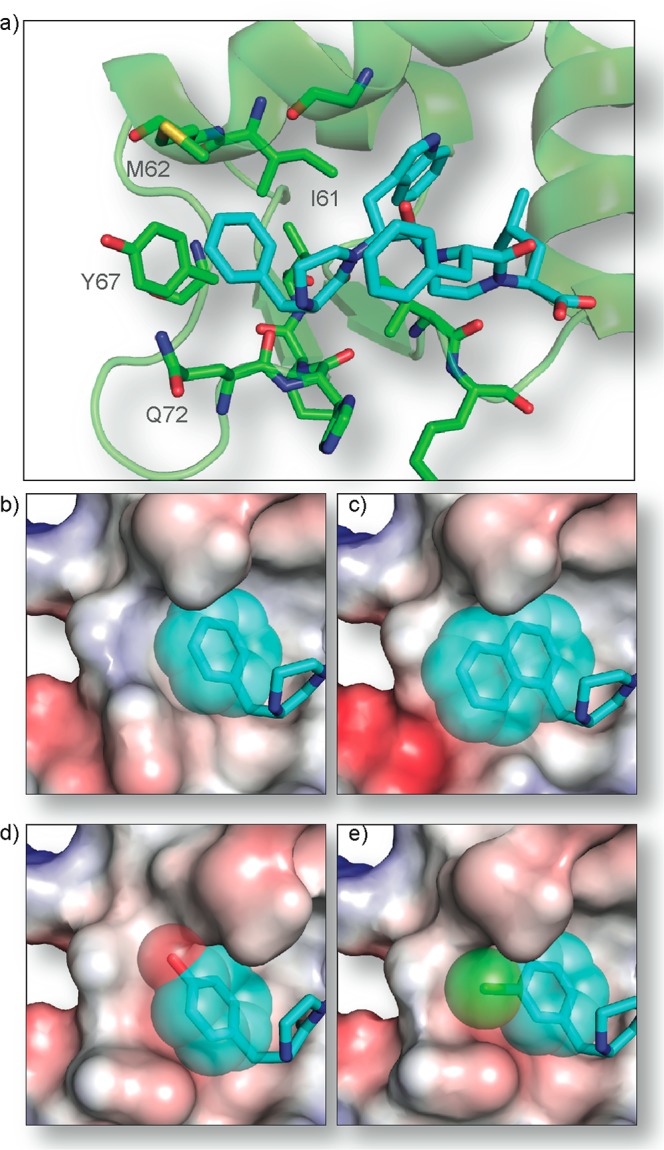
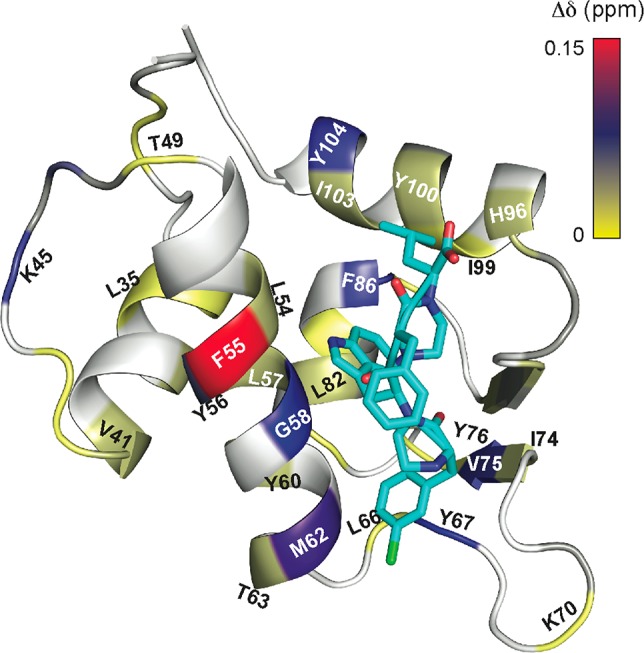
Protein-protein interactions encompass large surface areas, but often a handful of key residues dominate the binding energy landscape. Rationally designed small molecule scaffolds that reproduce the relative positioning and disposition of important binding residues, termed "hotspot residues", have been shown to successfully inhibit specific protein complexes. Although this strategy has led to development of novel synthetic inhibitors of protein complexes, often direct mimicry of natural amino acid residues does not lead to potent inhibitors. Experimental screening of focused compound libraries is used to further optimize inhibitors but the number of possible designs that can be efficiently synthesized and experimentally tested in academic settings is limited. We have applied the principles of computational protein design to optimization of nonpeptidic helix mimics as ligands for protein complexes. We describe the development of computational tools to design helix mimetics from canonical and noncanonical residue libraries and their application to two therapeutically important protein-protein interactions: p53-MDM2 and p300-HIF1α. The overall study provides a streamlined approach for discovering potent peptidomimetic inhibitors of protein-protein interactions.
蛋白质-蛋白质相互作用涉及较大的表面积,但通常少数关键残基主导着结合能格局。经合理设计的小分子支架能够重现重要结合残基(即“热点残基”)的相对位置和排布,已证明其可成功抑制特定的蛋白质复合物。尽管该策略已推动了蛋白质复合物新型合成抑制剂的开发,但对天然氨基酸残基的直接模拟往往无法得到强效抑制剂。聚焦化合物库的实验筛选用于进一步优化抑制剂,但在学术环境中能够有效合成并进行实验测试的可能设计数量有限。我们已将计算蛋白质设计原理应用于非肽类螺旋模拟物的优化,使其作为蛋白质复合物的配体。我们描述了从经典和非经典残基库设计螺旋模拟物的计算工具的开发及其在两种具有治疗重要性的蛋白质-蛋白质相互作用(p53-MDM2和p300-HIF1α)中的应用。总体研究为发现强效的蛋白质-蛋白质相互作用拟肽抑制剂提供了一种简化方法。