Berg Jeremy J, Coop Graham
Graduate Group in Population Biology, University of California, Davis, Davis, California, United States of America; Center for Population Biology, University of California, Davis, Davis, California, United States of America; Department of Evolution and Ecology, University of California, Davis, Davis, California, United States of America.
Center for Population Biology, University of California, Davis, Davis, California, United States of America; Department of Evolution and Ecology, University of California, Davis, Davis, California, United States of America.
PLoS Genet. 2014 Aug 7;10(8):e1004412. doi: 10.1371/journal.pgen.1004412. eCollection 2014 Aug.
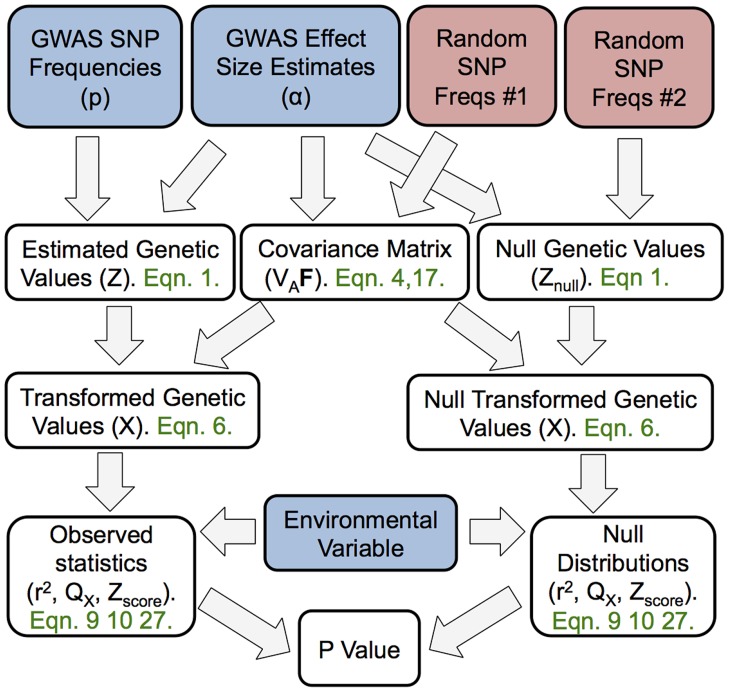
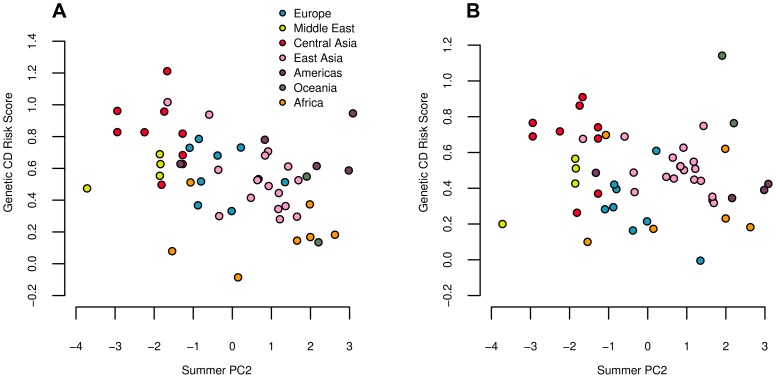
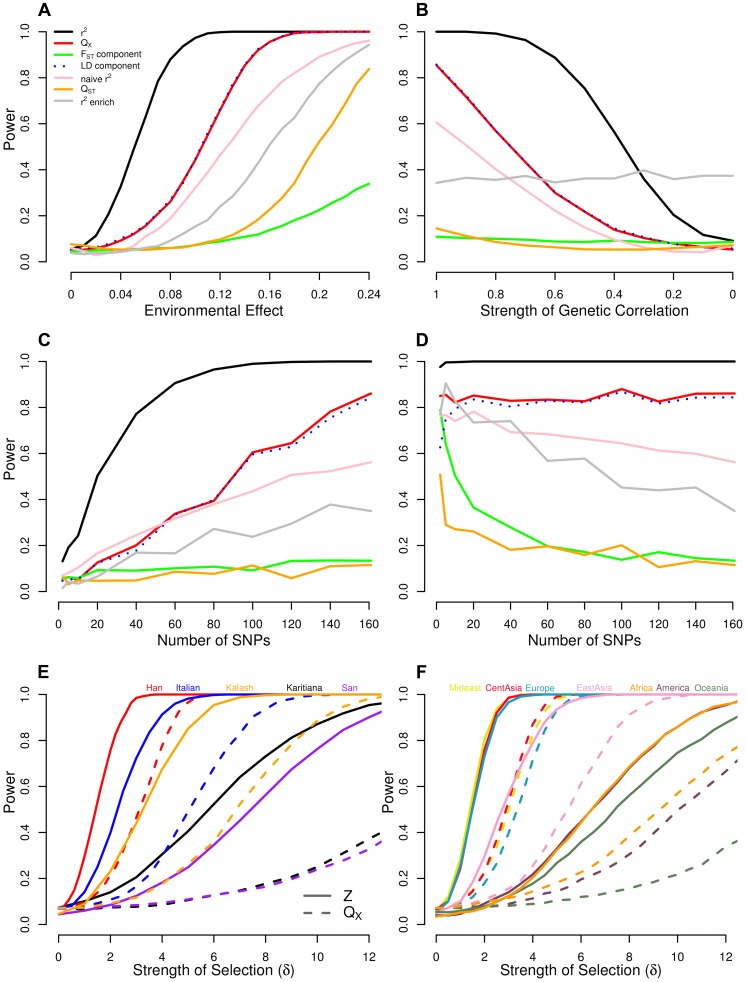
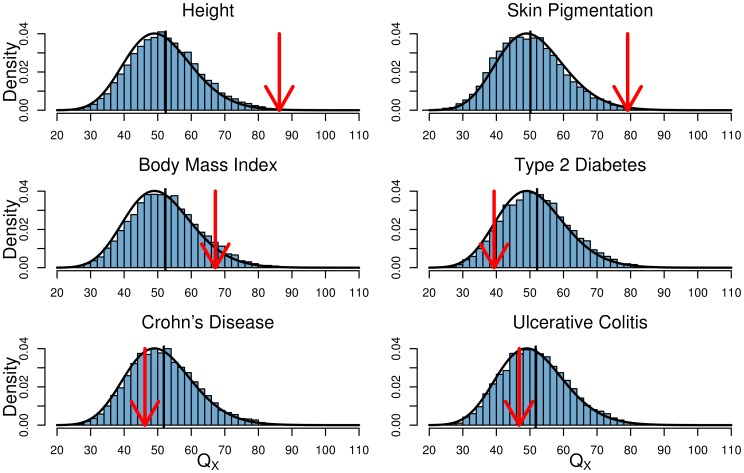
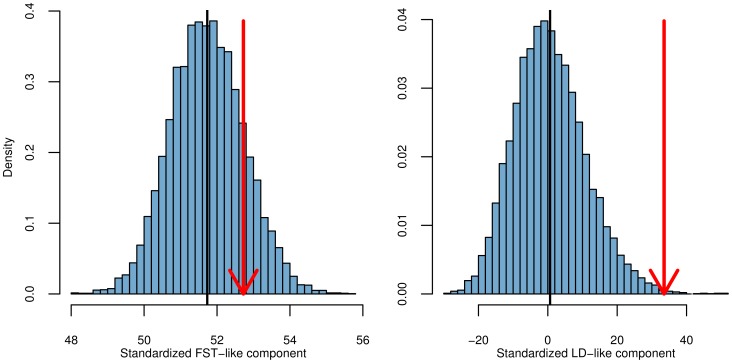
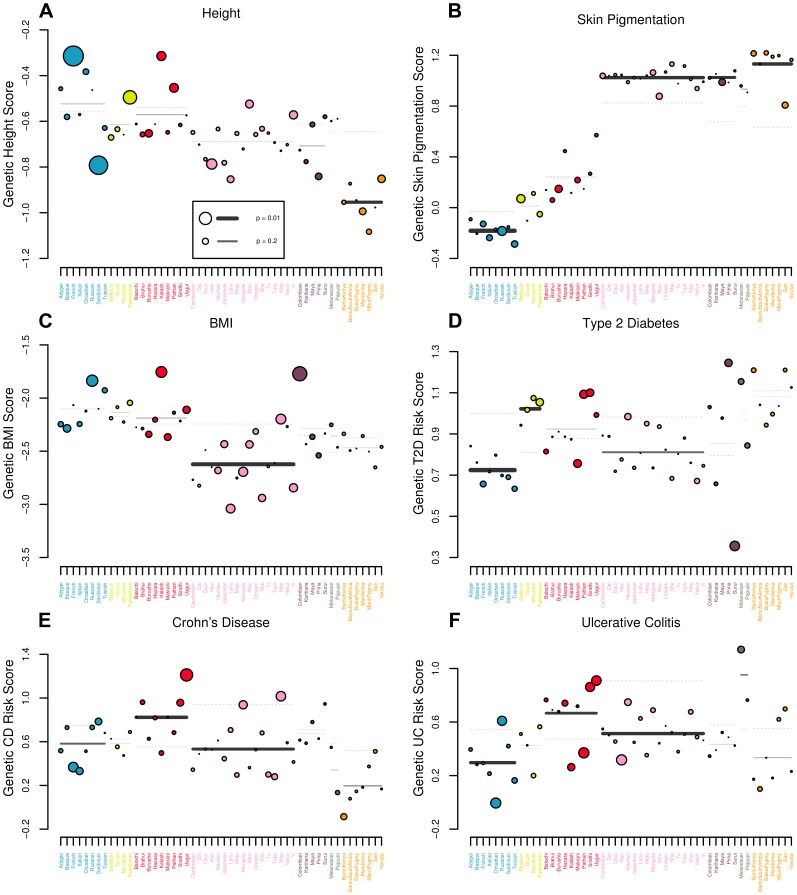
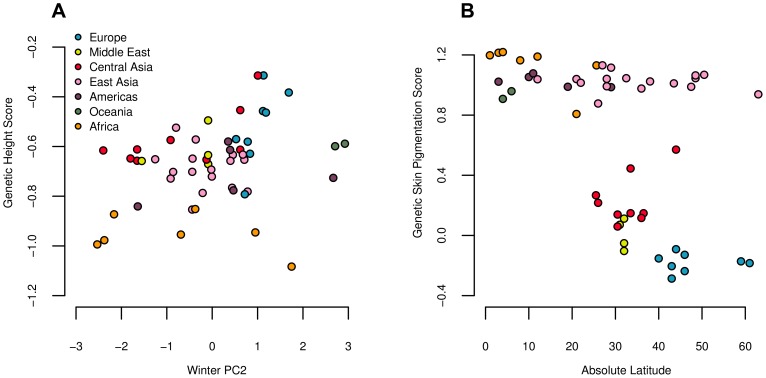
Adaptation in response to selection on polygenic phenotypes may occur via subtle allele frequencies shifts at many loci. Current population genomic techniques are not well posed to identify such signals. In the past decade, detailed knowledge about the specific loci underlying polygenic traits has begun to emerge from genome-wide association studies (GWAS). Here we combine this knowledge from GWAS with robust population genetic modeling to identify traits that may have been influenced by local adaptation. We exploit the fact that GWAS provide an estimate of the additive effect size of many loci to estimate the mean additive genetic value for a given phenotype across many populations as simple weighted sums of allele frequencies. We use a general model of neutral genetic value drift for an arbitrary number of populations with an arbitrary relatedness structure. Based on this model, we develop methods for detecting unusually strong correlations between genetic values and specific environmental variables, as well as a generalization of [Q(ST)/F(ST)] comparisons to test for over-dispersion of genetic values among populations. Finally we lay out a framework to identify the individual populations or groups of populations that contribute to the signal of overdispersion. These tests have considerably greater power than their single locus equivalents due to the fact that they look for positive covariance between like effect alleles, and also significantly outperform methods that do not account for population structure. We apply our tests to the Human Genome Diversity Panel (HGDP) dataset using GWAS data for height, skin pigmentation, type 2 diabetes, body mass index, and two inflammatory bowel disease datasets. This analysis uncovers a number of putative signals of local adaptation, and we discuss the biological interpretation and caveats of these results.
对多基因表型选择的适应性反应可能通过许多位点上微妙的等位基因频率变化而发生。当前的群体基因组技术并不适合识别此类信号。在过去十年中,全基因组关联研究(GWAS)已开始揭示有关多基因性状潜在特定位点的详细信息。在此,我们将GWAS的这些信息与强大的群体遗传模型相结合,以识别可能受到局部适应性影响的性状。我们利用GWAS提供的许多位点加性效应大小的估计值,将许多群体中给定表型的平均加性遗传值估计为等位基因频率的简单加权和。我们使用一个通用模型来描述具有任意亲缘结构的任意数量群体的中性遗传值漂移。基于此模型,我们开发了检测遗传值与特定环境变量之间异常强相关性的方法,以及[Q(ST)/F(ST)]比较的推广方法,以检验群体间遗传值的过度离散。最后,我们构建了一个框架来识别导致过度离散信号的单个群体或群体组。这些检验比其单一位点等效方法具有更大的功效,因为它们寻找类似效应等位基因之间的正协方差,并且也显著优于不考虑群体结构的方法。我们使用身高、皮肤色素沉着、2型糖尿病、体重指数的GWAS数据以及两个炎症性肠病数据集,将我们的检验应用于人类基因组多样性面板(HGDP)数据集。该分析揭示了许多局部适应性的推定信号,我们讨论了这些结果的生物学解释和注意事项。