Department of Computer Science and Engineering, Chinese University of Hong Kong, Hong Kong, China.
BMC Bioinformatics. 2014 Aug 27;15(1):291. doi: 10.1186/1471-2105-15-291.
State-of-the-art protein-ligand docking methods are generally limited by the traditionally low accuracy of their scoring functions, which are used to predict binding affinity and thus vital for discriminating between active and inactive compounds. Despite intensive research over the years, classical scoring functions have reached a plateau in their predictive performance. These assume a predetermined additive functional form for some sophisticated numerical features, and use standard multivariate linear regression (MLR) on experimental data to derive the coefficients.
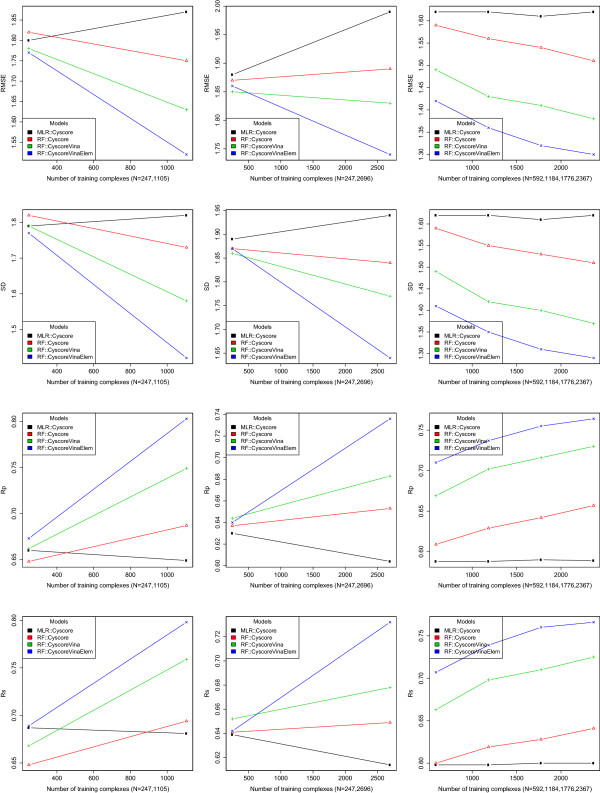
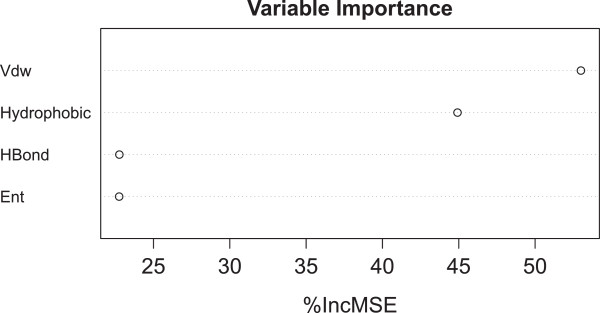
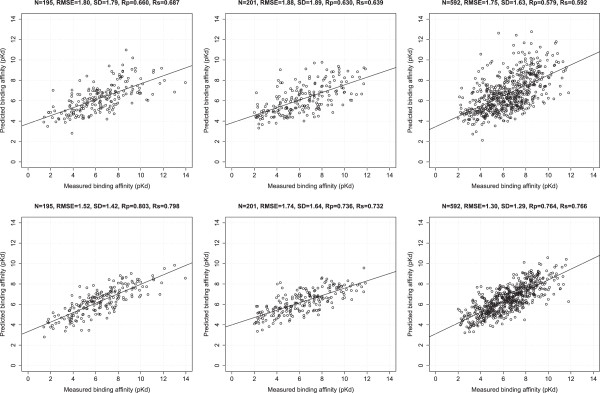
In this study we show that such a simple functional form is detrimental for the prediction performance of a scoring function, and replacing linear regression by machine learning techniques like random forest (RF) can improve prediction performance. We investigate the conditions of applying RF under various contexts and find that given sufficient training samples RF manages to comprehensively capture the non-linearity between structural features and measured binding affinities. Incorporating more structural features and training with more samples can both boost RF performance. In addition, we analyze the importance of structural features to binding affinity prediction using the RF variable importance tool. Lastly, we use Cyscore, a top performing empirical scoring function, as a baseline for comparison study.
Machine-learning scoring functions are fundamentally different from classical scoring functions because the former circumvents the fixed functional form relating structural features with binding affinities. RF, but not MLR, can effectively exploit more structural features and more training samples, leading to higher prediction performance. The future availability of more X-ray crystal structures will further widen the performance gap between RF-based and MLR-based scoring functions. This further stresses the importance of substituting RF for MLR in scoring function development.
最先进的蛋白质-配体对接方法通常受到其打分函数传统上准确性低的限制,打分函数用于预测结合亲和力,因此对于区分活性和非活性化合物至关重要。尽管多年来进行了密集的研究,但经典的打分函数在其预测性能方面已经达到了一个瓶颈。这些打分函数假设一些复杂的数值特征具有预定的加性函数形式,并使用标准多元线性回归(MLR)对实验数据进行分析以得出系数。
在本研究中,我们表明这种简单的函数形式不利于打分函数的预测性能,并且用机器学习技术(如随机森林(RF))替代线性回归可以提高预测性能。我们研究了在各种情况下应用 RF 的条件,并发现只要有足够的训练样本,RF 就能够全面捕捉结构特征与测量结合亲和力之间的非线性关系。纳入更多结构特征和使用更多样本进行训练都可以提高 RF 性能。此外,我们使用 RF 变量重要性工具分析结构特征对结合亲和力预测的重要性。最后,我们使用表现最佳的经验打分函数 Cyscore 作为比较研究的基线。
机器学习打分函数与经典打分函数在根本上不同,因为前者回避了将结构特征与结合亲和力联系起来的固定函数形式。RF 而不是 MLR 可以有效地利用更多的结构特征和更多的训练样本,从而提高预测性能。未来更多 X 射线晶体结构的可用性将进一步扩大 RF 基于和 MLR 基于打分函数之间的性能差距。这进一步强调了在打分函数开发中用 RF 替代 MLR 的重要性。