Ross Gregory A, Morris Garrett M, Biggin Philip C
Structural Bioinformatics and Computational Biochemistry, Department of Biochemistry, University of Oxford , South Parks Road, Oxford, Oxfordshire OX1 3QU, United Kingdom.
J Chem Theory Comput. 2013 Sep 10;9(9):4266-4274. doi: 10.1021/ct4004228. Epub 2013 Aug 5.
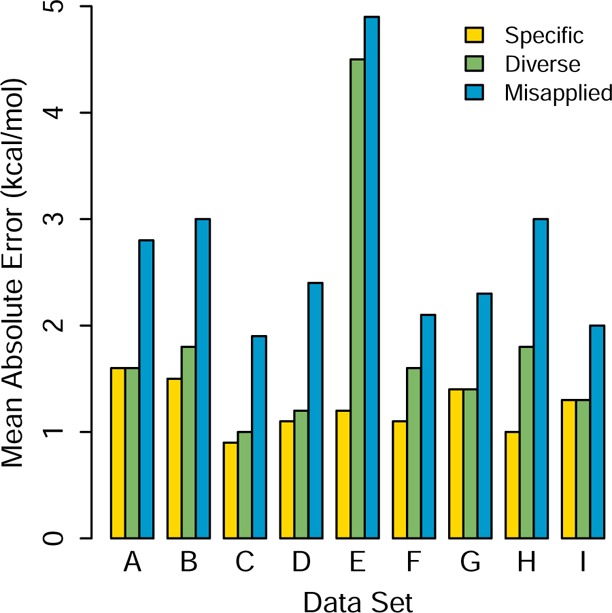
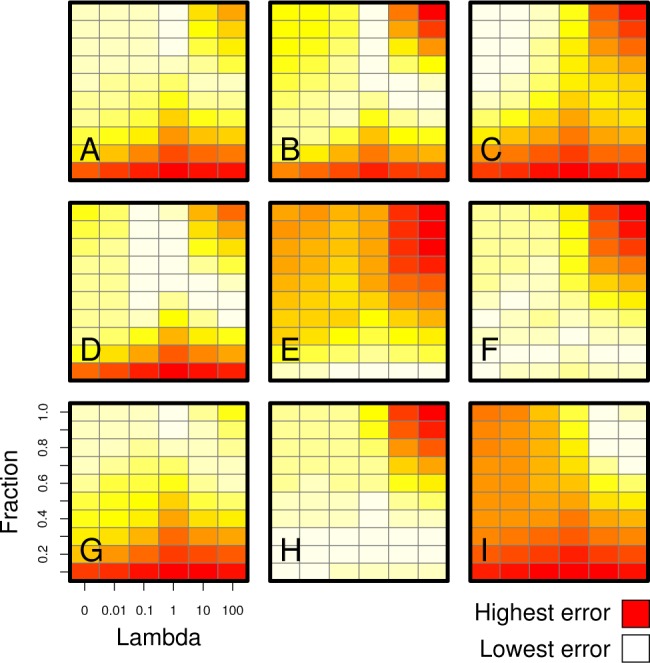
A major goal in computational chemistry has been to discover the set of rules that can accurately predict the binding affinity of any protein-drug complex, using only a single snapshot of its three-dimensional structure. Despite the continual development of structure-based models, predictive accuracy remains low, and the fundamental factors that inhibit the inference of all-encompassing rules have yet to be fully explored. Using statistical learning theory and information theory, here we prove that even the very best generalized structure-based model is inherently limited in its accuracy, and protein-specific models are always likely to be better. Our results refute the prevailing assumption that large data sets and advanced machine learning techniques will yield accurate, universally applicable models. We anticipate that the results will aid the development of more robust virtual screening strategies and scoring function error estimations.
计算化学的一个主要目标是发现一套规则,仅使用蛋白质-药物复合物三维结构的单个快照,就能准确预测其结合亲和力。尽管基于结构的模型不断发展,但预测准确性仍然很低,抑制推导全面规则的基本因素尚未得到充分探索。利用统计学习理论和信息论,我们在此证明,即使是最好的广义基于结构的模型,其准确性也存在固有局限性,特定于蛋白质的模型往往可能更好。我们的结果驳斥了一种普遍的假设,即大数据集和先进的机器学习技术将产生准确的、普遍适用的模型。我们预计这些结果将有助于开发更强大的虚拟筛选策略和评分函数误差估计。