Systems Biology and Mathematical Modeling Group, Max Planck Institute of Molecular Plant Physiology Potsdam-Golm, Germany.
Front Plant Sci. 2014 Sep 19;5:491. doi: 10.3389/fpls.2014.00491. eCollection 2014.
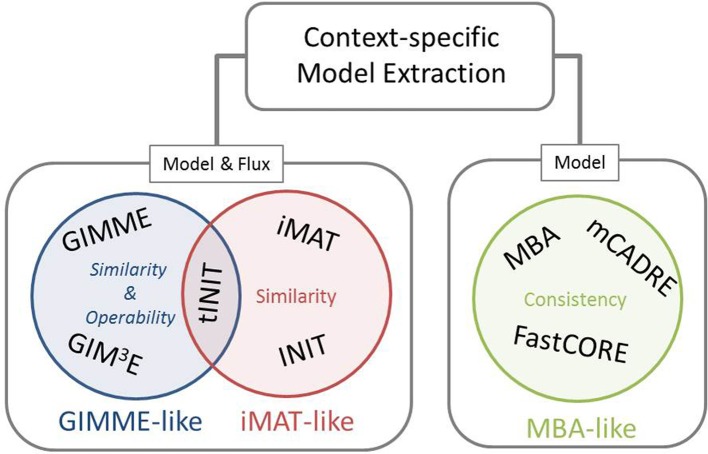
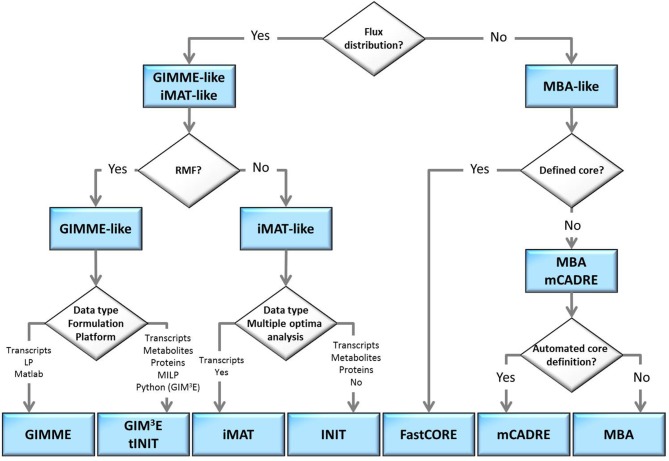
Genome-scale metabolic models (GEMs) are increasingly applied to investigate the physiology not only of simple prokaryotes, but also eukaryotes, such as plants, characterized with compartmentalized cells of multiple types. While genome-scale models aim at including the entirety of known metabolic reactions, mounting evidence has indicated that only a subset of these reactions is active in a given context, including: developmental stage, cell type, or environment. As a result, several methods have been proposed to reconstruct context-specific models from existing genome-scale models by integrating various types of high-throughput data. Here we present a mathematical framework that puts all existing methods under one umbrella and provides the means to better understand their functioning, highlight similarities and differences, and to help users in selecting a most suitable method for an application.
基因组规模代谢模型 (GEMs) 越来越多地被应用于研究生理机能,不仅针对简单的原核生物,也针对真核生物,如具有多种类型细胞区室化的植物。尽管基因组规模模型旨在包含所有已知的代谢反应,但越来越多的证据表明,在给定的环境中,只有这些反应的一部分是活跃的,包括:发育阶段、细胞类型或环境。因此,已经提出了几种方法,通过整合各种类型的高通量数据,从现有的基因组规模模型中重建特定于上下文的模型。在这里,我们提出了一个数学框架,将所有现有的方法都纳入一个框架中,并提供了更好地理解它们的功能、突出相似之处和不同之处的手段,以及帮助用户为应用选择最合适的方法。