Parker Hilary S, Corrada Bravo Héctor, Leek Jeffrey T
Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health , Baltimore, MD , USA.
Center for Bioinformatics and Computational Biology, Department of Computer Science, University of Maryland , College Park, MD , USA.
PeerJ. 2014 Sep 23;2:e561. doi: 10.7717/peerj.561. eCollection 2014.
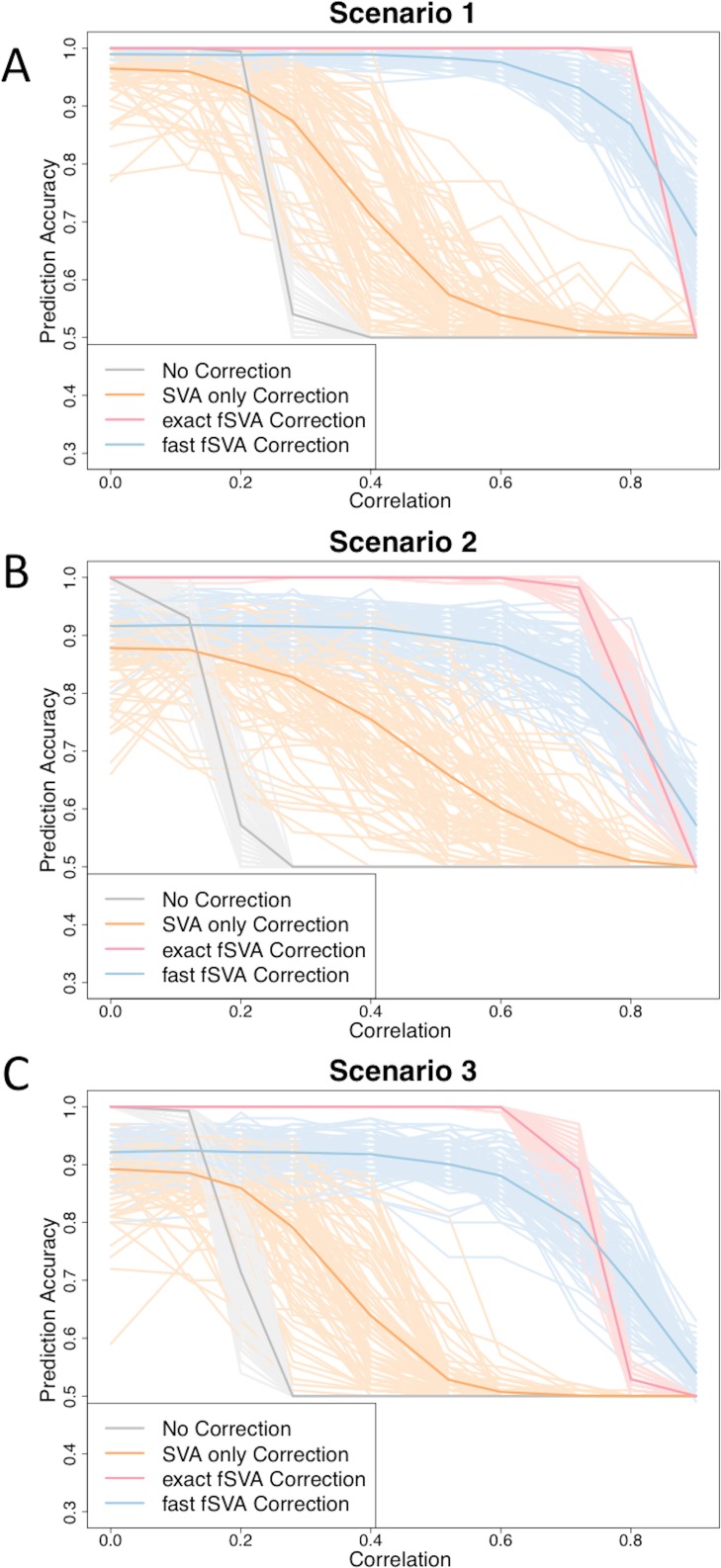
Batch effects are responsible for the failure of promising genomic prognostic signatures, major ambiguities in published genomic results, and retractions of widely-publicized findings. Batch effect corrections have been developed to remove these artifacts, but they are designed to be used in population studies. But genomic technologies are beginning to be used in clinical applications where samples are analyzed one at a time for diagnostic, prognostic, and predictive applications. There are currently no batch correction methods that have been developed specifically for prediction. In this paper, we propose an new method called frozen surrogate variable analysis (fSVA) that borrows strength from a training set for individual sample batch correction. We show that fSVA improves prediction accuracy in simulations and in public genomic studies. fSVA is available as part of the sva Bioconductor package.
批次效应导致了有前景的基因组预后特征失效、已发表基因组结果中的重大歧义以及广泛宣传的研究结果被撤回。已经开发了批次效应校正方法来消除这些假象,但它们是设计用于人群研究的。然而,基因组技术正开始用于临床应用,在这些应用中,样本是一次一个地进行分析以用于诊断、预后和预测。目前还没有专门为预测开发的批次校正方法。在本文中,我们提出了一种名为冻结替代变量分析(fSVA)的新方法,该方法从训练集中汲取力量以进行单个样本的批次校正。我们表明,fSVA在模拟和公共基因组研究中提高了预测准确性。fSVA作为sva Bioconductor软件包的一部分可用。