Jaffe Andrew E, Hyde Thomas, Kleinman Joel, Weinbergern Daniel R, Chenoweth Joshua G, McKay Ronald D, Leek Jeffrey T, Colantuoni Carlo
Lieber Institute for Brain Development, 855 N Wolfe St, Ste 300, Baltimore, MD, 21205, USA.
Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, 615 N Wolfe St, Baltimore, MD, 21205, USA.
BMC Bioinformatics. 2015 Nov 6;16:372. doi: 10.1186/s12859-015-0808-5.
Genomic data production is at its highest level and continues to increase, making available novel primary data and existing public data to researchers for exploration. Here we explore the consequences of "batch" correction for biological discovery in two publicly available expression datasets. We consider this to include the estimation of and adjustment for wide-spread systematic heterogeneity in genomic measurements that is unrelated to the effects under study, whether it be technical or biological in nature.
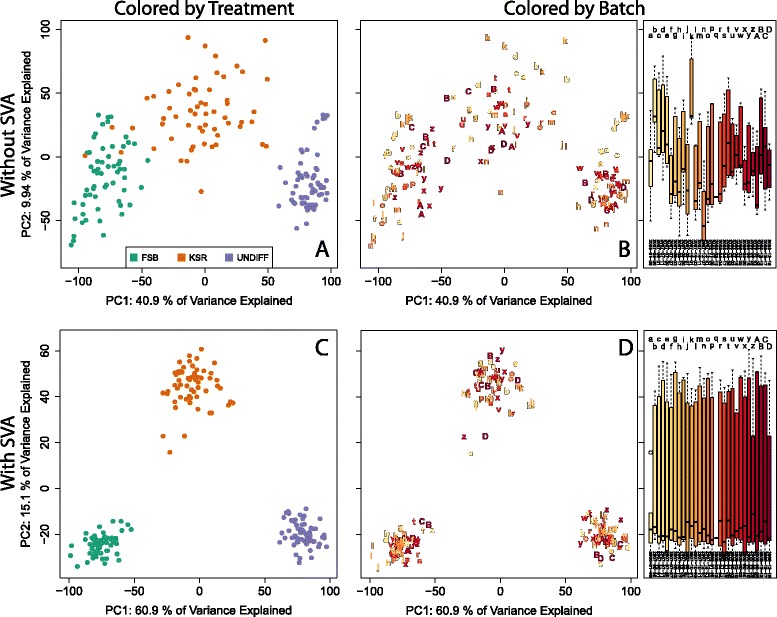
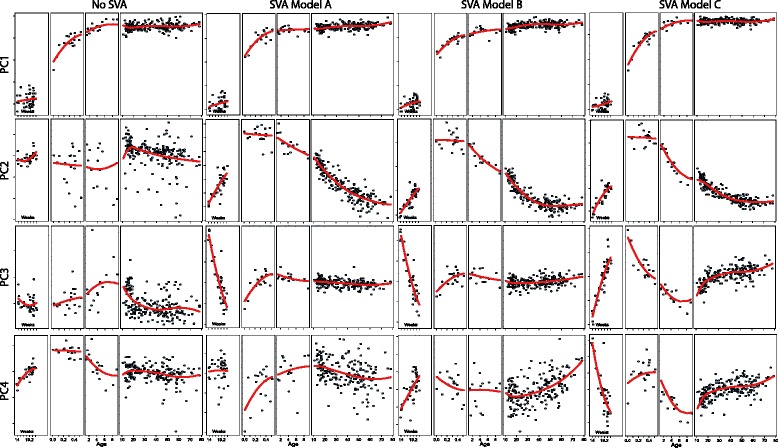
We present three illustrative data analyses using surrogate variable analysis (SVA) and describe how to perform artifact discovery in light of natural heterogeneity within biological groups, secondary biological questions of interest, and non-linear treatment effects in a dataset profiling differentiating pluripotent cells (GSE32923) and another from human brain tissue (GSE30272).
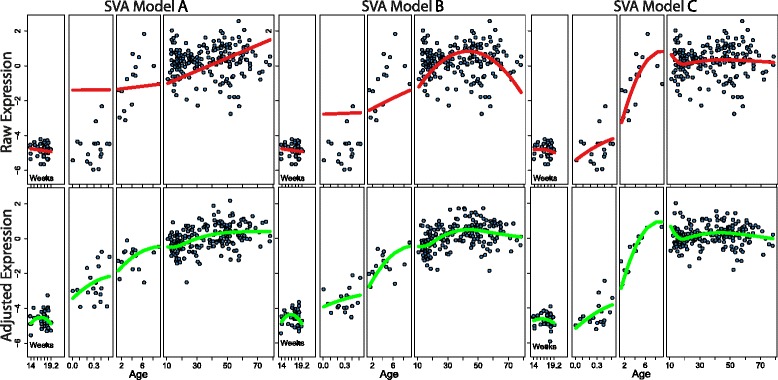
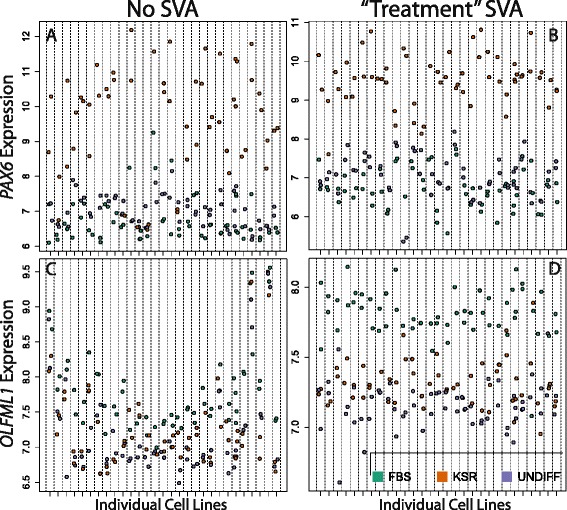
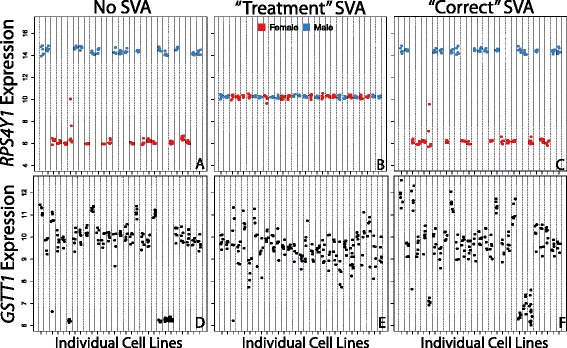
Careful specification of biological effects of interest is very important to factor-based approaches like SVA. We demonstrate greatly sharpened global and gene-specific differential expression across treatment groups in stem cell systems. Similarly, we demonstrate how to preserve major non-linear effects of age across the lifespan in the brain dataset. However, the gains in precisely defining known effects of interest come at the cost of much other information in the "cleaned" data, including sex, common copy number effects and sample or cell line-specific molecular behavior.
Our analyses indicate that data "cleaning" can be an important component of high-throughput genomic data analysis when interrogating explicitly defined effects in the context of data affected by robust technical artifacts. However, caution should be exercised to avoid removing biological signal of interest. It is also important to note that open data exploration is not possible after such supervised "cleaning", because effects beyond those stipulated by the researcher may have been removed. With the goal of making these statistical algorithms more powerful and transparent to researchers in the biological sciences, we provide exploratory plots and accompanying R code for identifying and guiding "cleaning" process (https://github.com/andrewejaffe/StemCellSVA). The impact of these methods is significant enough that we have made newly processed data available for the brain data set at http://braincloud.jhmi.edu/plots/ and GSE30272.
基因组数据的产出处于最高水平且持续增长,为研究人员提供了新的原始数据和现有的公共数据以供探索。在此,我们在两个公开可用的表达数据集中探讨“批次”校正对生物学发现的影响。我们认为这包括对基因组测量中广泛存在的系统异质性进行估计和调整,这种异质性与所研究的效应无关,无论其本质是技术方面还是生物学方面。
我们展示了三项使用替代变量分析(SVA)的说明性数据分析,并描述了如何根据生物组内的自然异质性、感兴趣的次要生物学问题以及在一个区分多能细胞的数据集(GSE32923)和另一个来自人类脑组织的数据集(GSE30272)中的非线性处理效应来进行伪像发现。
仔细确定感兴趣的生物学效应对于像SVA这样基于因素的方法非常重要。我们展示了在干细胞系统中,跨治疗组的全局和基因特异性差异表达大大增强。同样,我们展示了如何在脑数据集中保留年龄在整个生命周期中的主要非线性效应。然而,精确界定已知感兴趣效应所带来的收获是以“清理后”数据中的许多其他信息为代价的,包括性别、常见的拷贝数效应以及样本或细胞系特异性的分子行为。
我们的分析表明,在受强大技术伪像影响的数据背景下询问明确界定的效应时,数据“清理”可以是高通量基因组数据分析的一个重要组成部分。然而,应谨慎行事以避免去除感兴趣的生物学信号。还需注意的是,在这种有监督的“清理”之后,开放数据探索是不可能的,因为研究人员规定之外的效应可能已被去除。为了使这些统计算法对生物科学领域的研究人员更强大且更透明,我们提供了探索性图表以及用于识别和指导“清理”过程的配套R代码(https://github.com/andrewejaffe/StemCellSVA)。这些方法的影响足够显著,以至于我们已在http://braincloud.jhmi.edu/plots/和GSE30272上提供了脑数据集的新处理后数据。