Haugum Kjersti, Johansen Jostein, Gabrielsen Christina, Brandal Lin T, Bergh Kåre, Ussery David W, Drabløs Finn, Afset Jan Egil
Department of Laboratory Medicine, Children's and Women's Health, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway.
Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway.
PLoS One. 2014 Oct 31;9(10):e111788. doi: 10.1371/journal.pone.0111788. eCollection 2014.
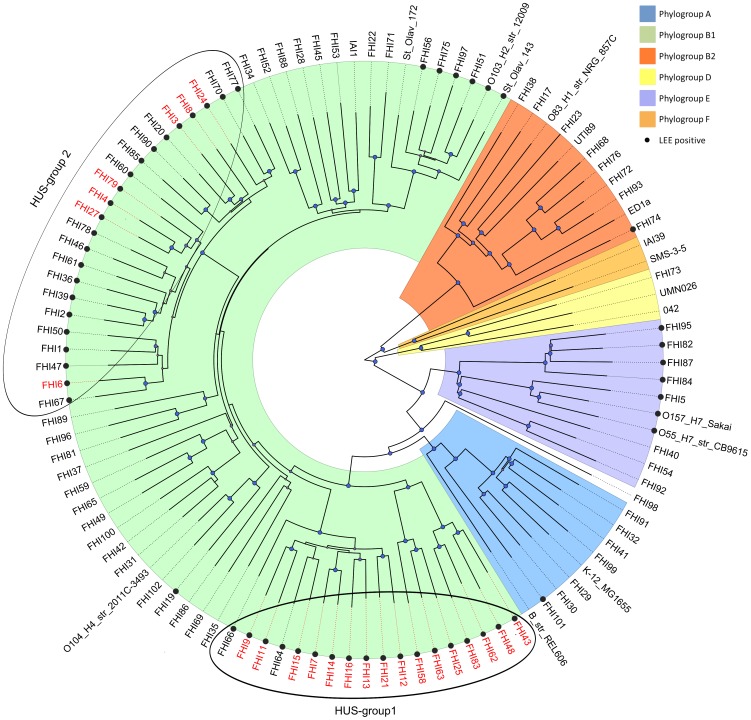
Shiga toxin-producing Escherichia coli (STEC) cause infections in humans ranging from asymptomatic carriage to bloody diarrhoea and haemolytic uremic syndrome (HUS). Here we present whole genome comparison of Norwegian non-O157 STEC strains with the aim to distinguish between strains with the potential to cause HUS and less virulent strains. Whole genome sequencing and comparisons were performed across 95 non-O157 STEC strains. Twenty-three of these were classified as HUS-associated, including strains from patients with HUS (n = 19) and persons with an epidemiological link to a HUS-case (n = 4). Genomic comparison revealed considerable heterogeneity in gene content across the 95 STEC strains. A clear difference in gene profile was observed between strains with and without the Locus of Enterocyte Effacement (LEE) pathogenicity island. Phylogenetic analysis of the core genome showed high degree of diversity among the STEC strains, but all HUS-associated STEC strains were distributed in two distinct clusters within phylogroup B1. However, non-HUS strains were also found in these clusters. A number of accessory genes were found to be significantly overrepresented among HUS-associated STEC, but none of them were unique to this group of strains, suggesting that different sets of genes may contribute to the pathogenic potential in different phylogenetic STEC lineages. In this study we were not able to clearly distinguish between HUS-associated and non-HUS non-O157 STEC by extensive genome comparisons. Our results indicate that STECs from different phylogenetic backgrounds have independently acquired virulence genes that determine pathogenic potential, and that the content of such genes is overlapping between HUS-associated and non-HUS strains.
产志贺毒素大肠杆菌(STEC)可导致人类感染,症状从无症状携带到血性腹泻和溶血尿毒综合征(HUS)不等。在此,我们展示了挪威非O157 STEC菌株的全基因组比较,目的是区分有导致HUS潜力的菌株和毒性较低的菌株。对95株非O157 STEC菌株进行了全基因组测序和比较。其中23株被归类为与HUS相关,包括来自HUS患者的菌株(n = 19)和与HUS病例有流行病学关联的人员的菌株(n = 4)。基因组比较显示,95株STEC菌株的基因内容存在相当大的异质性。在有和没有肠细胞脱落位点(LEE)致病岛的菌株之间观察到基因谱的明显差异。核心基因组的系统发育分析表明,STEC菌株之间存在高度多样性,但所有与HUS相关的STEC菌株都分布在B1系统发育群内的两个不同簇中。然而,在这些簇中也发现了非HUS菌株。发现一些辅助基因在与HUS相关的STEC中显著富集,但没有一个是该组菌株所特有的,这表明不同的基因集可能在不同的系统发育STEC谱系中促成致病潜力。在本研究中,我们无法通过广泛的基因组比较清楚地区分与HUS相关和非HUS的非O157 STEC。我们的结果表明,来自不同系统发育背景的STEC独立获得了决定致病潜力的毒力基因,并且这些基因的内容在与HUS相关和非HUS菌株之间重叠。