Pierlé Sebastián Aguilar, Rosshandler Ivan Imaz, Kerudin Ammielle Akim, Sambono Jacqueline, Lew-Tabor Ala, Rolls Peter, Rangel-Escareño Claudia, Brayton Kelly A
Program in Genomics, Department of Veterinary Microbiology and Pathology, Paul G. Allen School for Global Animal Health, Washington State University, Pullman, WA 99164-7040, USA.
National Institute of Genomic Medicine, Computational Genomics Lab, Mexico City 14610, Mexico.
Pathogens. 2014 Jan 14;3(1):57-72. doi: 10.3390/pathogens3010057.
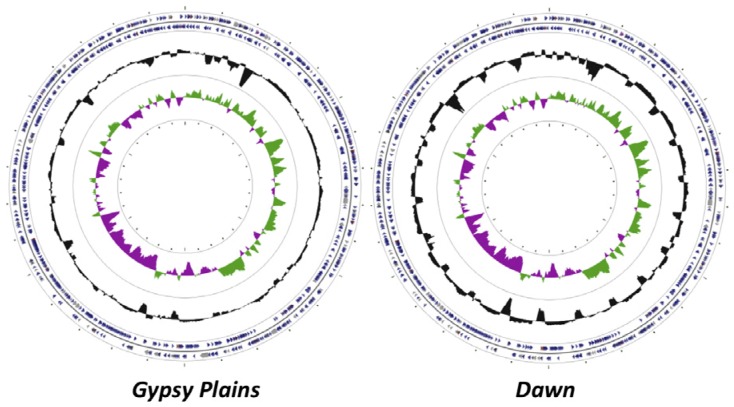
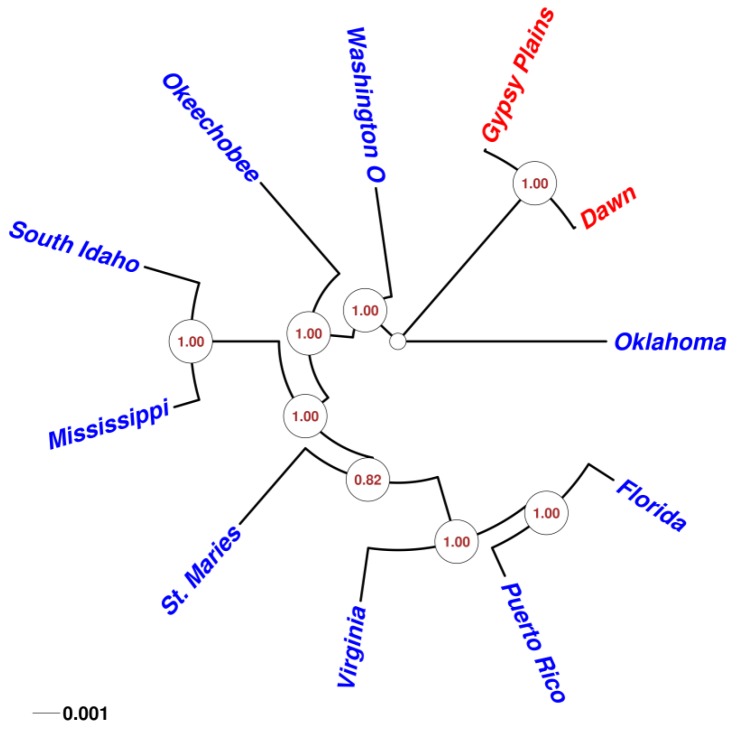
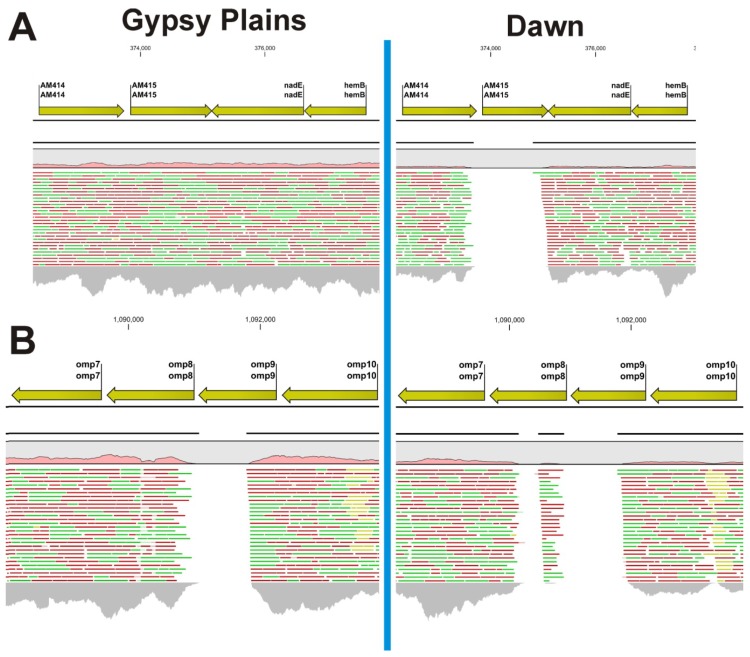
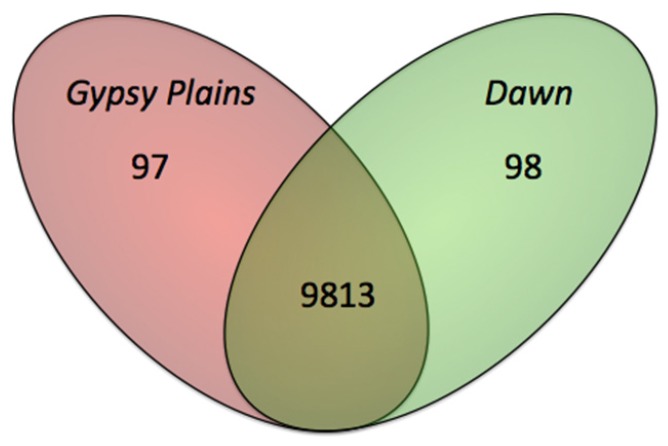
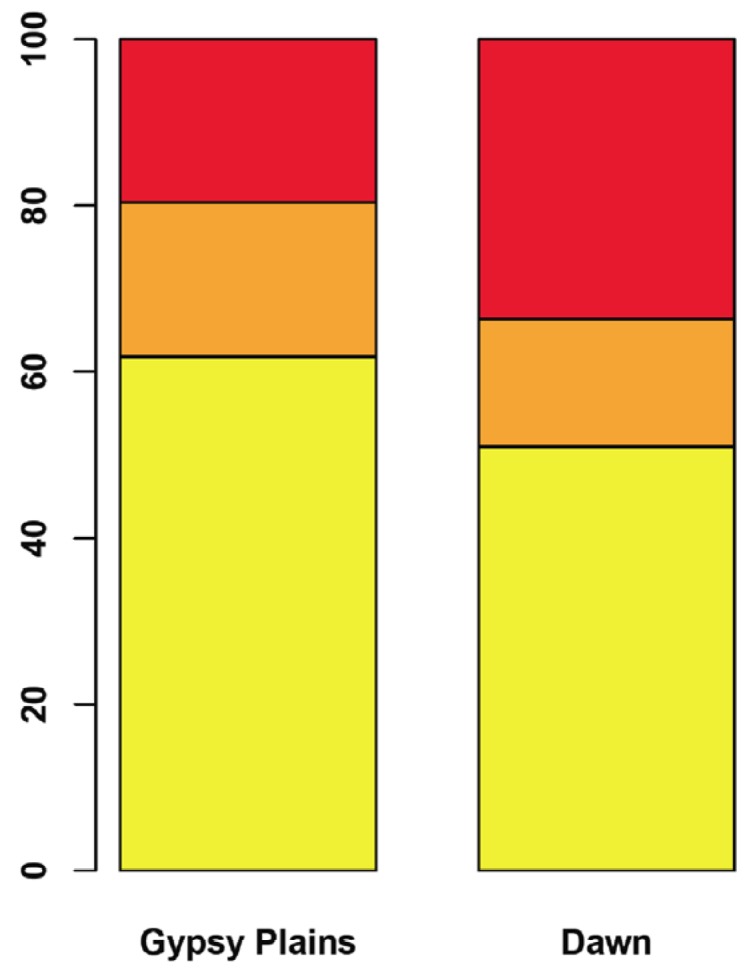
The ability to capture genetic variation with unprecedented resolution improves our understanding of bacterial populations and their ability to cause disease. The goal of the pathogenomics era is to define genetic diversity that results in disease. Despite the economic losses caused by vector-borne bacteria in the Order Rickettsiales, little is known about the genetic variants responsible for observed phenotypes. The tick-transmitted rickettsial pathogen infects cattle in tropical and subtropical regions worldwide, including Australia. Genomic analysis of North American strains reveals a closed core genome defined by high levels of Single Nucleotide Polymorphisms (SNPs). Here we report the first genome sequences and comparative analysis for Australian strains that differ in virulence and transmissibility. A list of genetic differences that segregate with phenotype was evaluated for the ability to distinguish the attenuated strain from virulent field strains. Phylogenetic analyses of the Australian strains revealed a marked evolutionary distance from all previously sequenced strains. SNP analysis showed a strikingly reduced genetic diversity between these strains, with the smallest number of SNPs detected between any two strains. The low diversity between these phenotypically distinct bacteria presents a unique opportunity to identify the genetic determinants of virulence and transmission.
以前所未有的分辨率捕获遗传变异的能力,增进了我们对细菌群体及其致病能力的理解。病原体基因组学时代的目标是定义导致疾病的遗传多样性。尽管立克次氏体目中由媒介传播的细菌造成了经济损失,但对于导致观察到的表型的遗传变异却知之甚少。蜱传播的立克次氏体病原体感染包括澳大利亚在内的全球热带和亚热带地区的牛。对北美菌株的基因组分析揭示了一个由高水平单核苷酸多态性(SNP)定义的封闭核心基因组。在此,我们报告了澳大利亚菌株的首批基因组序列及比较分析,这些菌株在毒力和传播性方面存在差异。对与表型相关的遗传差异列表进行了评估,以确定其区分减毒株和强毒野外菌株的能力。对澳大利亚菌株的系统发育分析显示,与所有先前测序的菌株存在显著的进化距离。SNP分析表明,这些菌株之间的遗传多样性显著降低,任意两个菌株之间检测到的SNP数量最少。这些表型不同的细菌之间的低多样性为鉴定毒力和传播的遗传决定因素提供了独特的机会。