James Kevin A, Verkhivker Gennady M
School of Computational Sciences and Crean School of Health and Life Sciences, Schmid College of Science and Technology, Chapman University, Orange, California, United States of America.
School of Computational Sciences and Crean School of Health and Life Sciences, Schmid College of Science and Technology, Chapman University, Orange, California, United States of America; Department of Pharmacology, University of California San Diego, La Jolla, California, United States of America.
PLoS One. 2014 Nov 26;9(11):e113488. doi: 10.1371/journal.pone.0113488. eCollection 2014.
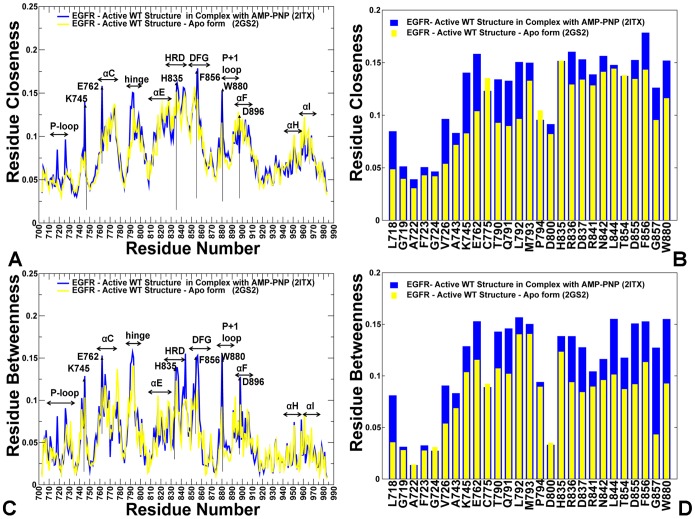
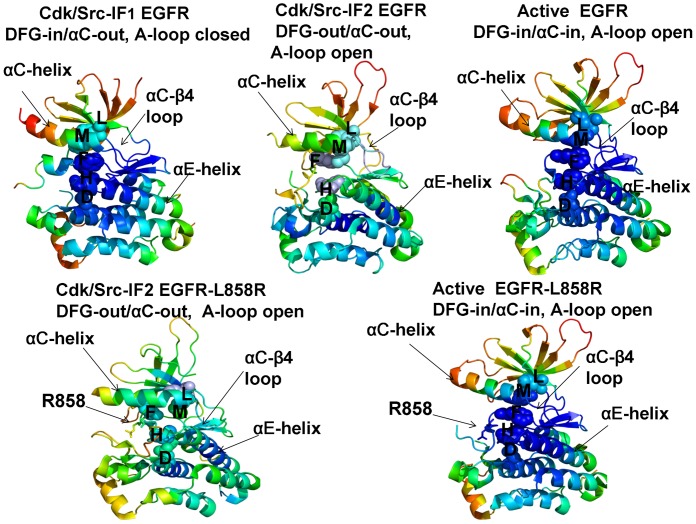
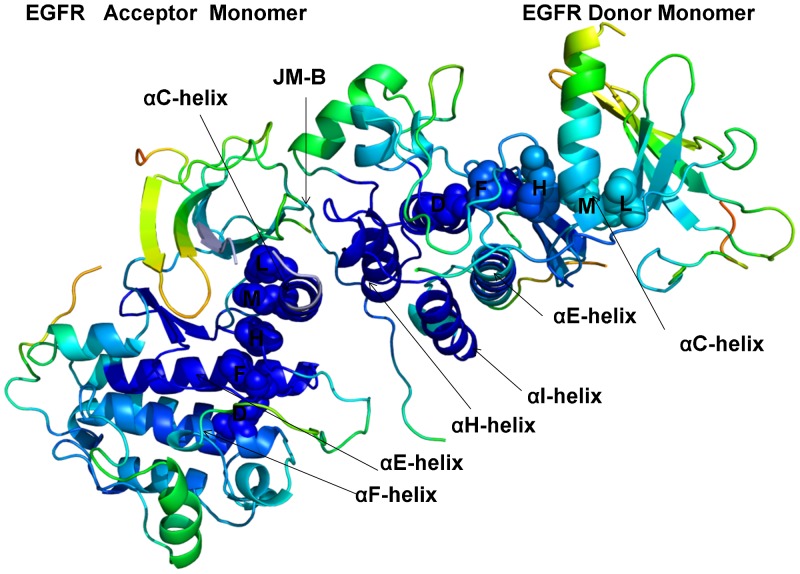
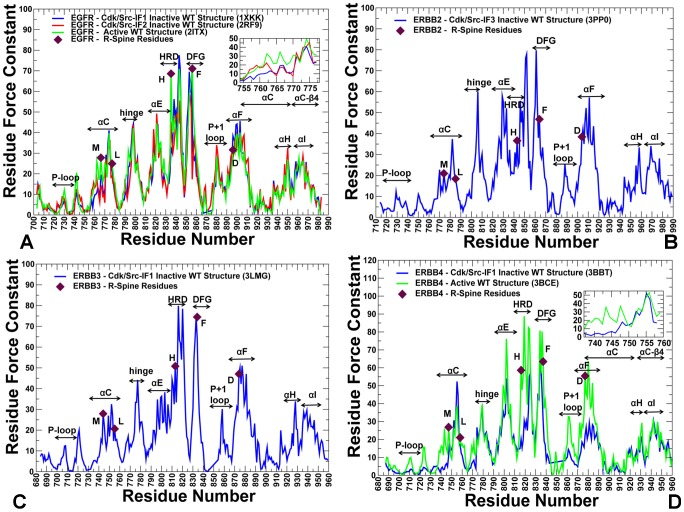
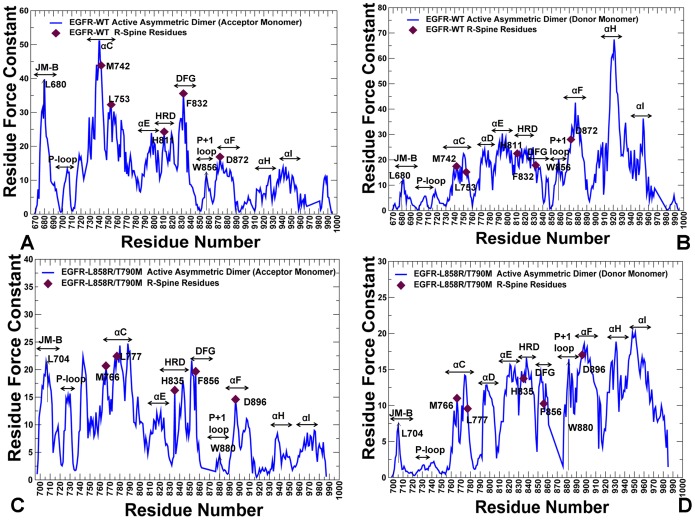
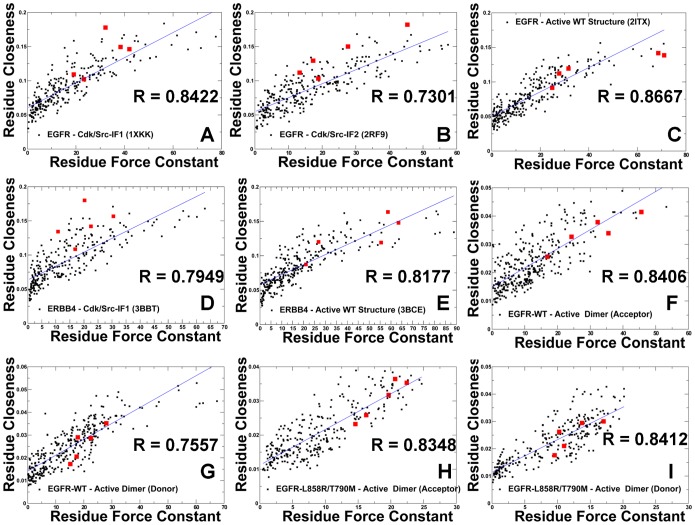
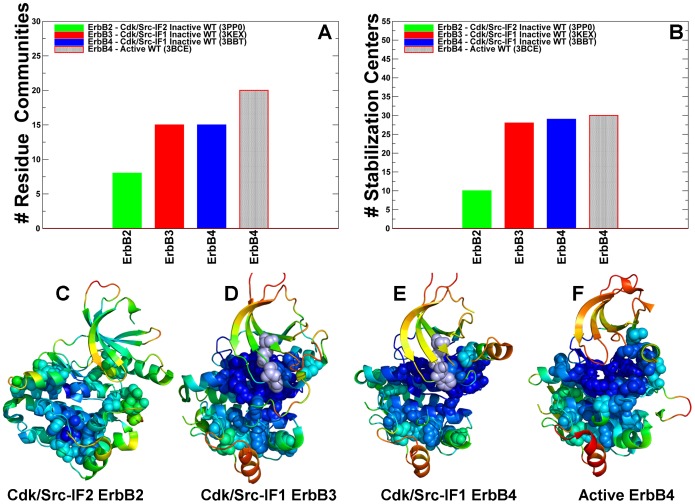
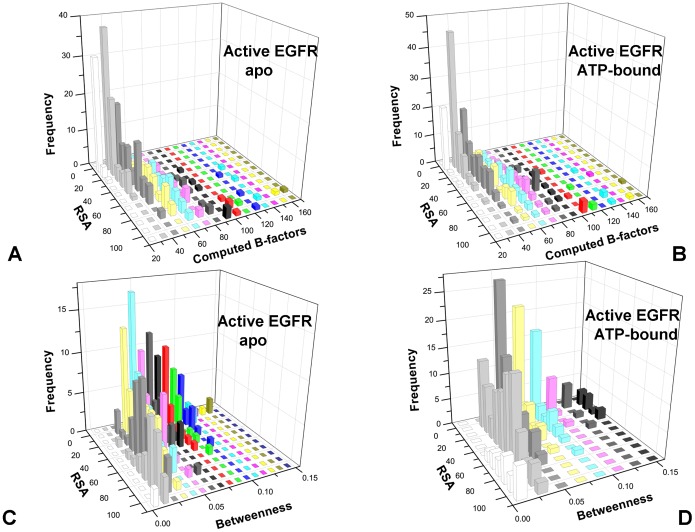
The ErbB protein tyrosine kinases are among the most important cell signaling families and mutation-induced modulation of their activity is associated with diverse functions in biological networks and human disease. We have combined molecular dynamics simulations of the ErbB kinases with the protein structure network modeling to characterize the reorganization of the residue interaction networks during conformational equilibrium changes in the normal and oncogenic forms. Structural stability and network analyses have identified local communities integrated around high centrality sites that correspond to the regulatory spine residues. This analysis has provided a quantitative insight to the mechanism of mutation-induced "superacceptor" activity in oncogenic EGFR dimers. We have found that kinase activation may be determined by allosteric interactions between modules of structurally stable residues that synchronize the dynamics in the nucleotide binding site and the αC-helix with the collective motions of the integrating αF-helix and the substrate binding site. The results of this study have pointed to a central role of the conserved His-Arg-Asp (HRD) motif in the catalytic loop and the Asp-Phe-Gly (DFG) motif as key mediators of structural stability and allosteric communications in the ErbB kinases. We have determined that residues that are indispensable for kinase regulation and catalysis often corresponded to the high centrality nodes within the protein structure network and could be distinguished by their unique network signatures. The optimal communication pathways are also controlled by these nodes and may ensure efficient allosteric signaling in the functional kinase state. Structure-based network analysis has quantified subtle effects of ATP binding on conformational dynamics and stability of the EGFR structures. Consistent with the NMR studies, we have found that nucleotide-induced modulation of the residue interaction networks is not limited to the ATP site, and may enhance allosteric cooperativity with the substrate binding region by increasing communication capabilities of mediating residues.
表皮生长因子受体(ErbB)蛋白酪氨酸激酶是最重要的细胞信号传导家族之一,其活性的突变诱导调节与生物网络和人类疾病中的多种功能相关。我们将ErbB激酶的分子动力学模拟与蛋白质结构网络建模相结合,以表征正常和致癌形式的构象平衡变化过程中残基相互作用网络的重组。结构稳定性和网络分析确定了围绕与调节脊柱残基相对应的高中心性位点整合的局部群落。该分析为致癌性表皮生长因子受体(EGFR)二聚体中突变诱导的“超级受体”活性机制提供了定量见解。我们发现激酶激活可能由结构稳定残基模块之间的变构相互作用决定,这些相互作用使核苷酸结合位点和αC螺旋中的动力学与整合的αF螺旋和底物结合位点的集体运动同步。这项研究的结果表明,催化环中保守的组氨酸 - 精氨酸 - 天冬氨酸(HRD)基序和天冬氨酸 - 苯丙氨酸 - 甘氨酸(DFG)基序在ErbB激酶的结构稳定性和变构通讯中起关键介导作用。我们确定,激酶调节和催化必不可少的残基通常对应于蛋白质结构网络中的高中心性节点,并可通过其独特的网络特征加以区分。最佳通讯途径也由这些节点控制,并可确保功能性激酶状态下的有效变构信号传导。基于结构的网络分析量化了ATP结合对EGFR结构构象动力学和稳定性的微妙影响。与核磁共振研究一致,我们发现核苷酸诱导的残基相互作用网络调节不仅限于ATP位点,还可能通过增加介导残基的通讯能力来增强与底物结合区域的变构协同作用。