BMC Bioinformatics. 2014;15 Suppl 15(Suppl 15):S3. doi: 10.1186/1471-2105-15-S15-S3. Epub 2014 Dec 3.
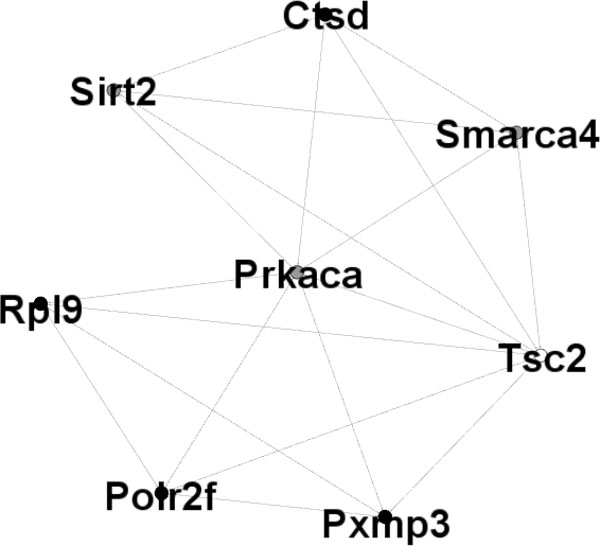
Differential coexpression analysis usually requires the definition of 'distance' or 'similarity' between measured datasets. Until now, the most common choice is Pearson correlation coefficient. However, Pearson correlation coefficient is sensitive to outliers. Biweight midcorrelation is considered to be a good alternative to Pearson correlation since it is more robust to outliers. In this paper, we introduce to use Biweight Midcorrelation to measure 'similarity' between gene expression profiles, and provide a new approach for gene differential coexpression analysis. Firstly, we calculate the biweight midcorrelation coefficients between all gene pairs. Then, we filter out non-informative correlation pairs using the 'half-thresholding' strategy and calculate the differential coexpression value of gene, The experimental results on simulated data show that the new approach performed better than three previously published differential coexpression analysis (DCEA) methods. Moreover, we use the maximum clique analysis to gene subset included genes identified by our approach and previously reported T2D-related genes, many additional discoveries can be found through our method.
差异共表达分析通常需要定义测量数据集之间的“距离”或“相似性”。到目前为止,最常见的选择是皮尔逊相关系数。然而,皮尔逊相关系数对异常值很敏感。双权中位数相关被认为是皮尔逊相关的一个很好的替代品,因为它对异常值更稳健。在本文中,我们引入了使用双权中位数相关来衡量基因表达谱之间的“相似性”,并提供了一种新的基因差异共表达分析方法。首先,我们计算所有基因对之间的双权中位数相关系数。然后,我们使用“半阈值”策略过滤掉非信息相关的相关对,并计算基因的差异共表达值。模拟数据的实验结果表明,该新方法的性能优于三种先前发表的差异共表达分析(DCEA)方法。此外,我们使用最大团分析对我们的方法和先前报道的 T2D 相关基因识别的基因子集进行分析,通过我们的方法可以发现许多额外的发现。