Wallace Jason G, Bradbury Peter J, Zhang Nengyi, Gibon Yves, Stitt Mark, Buckler Edward S
Institute for Genomic Diversity, Cornell University, Ithaca, New York, United States of America.
Institute for Genomic Diversity, Cornell University, Ithaca, New York, United States of America; United States Department of Agriculture-Agricultural Research Service, Ithaca, New York, United States of America.
PLoS Genet. 2014 Dec 4;10(12):e1004845. doi: 10.1371/journal.pgen.1004845. eCollection 2014 Dec.
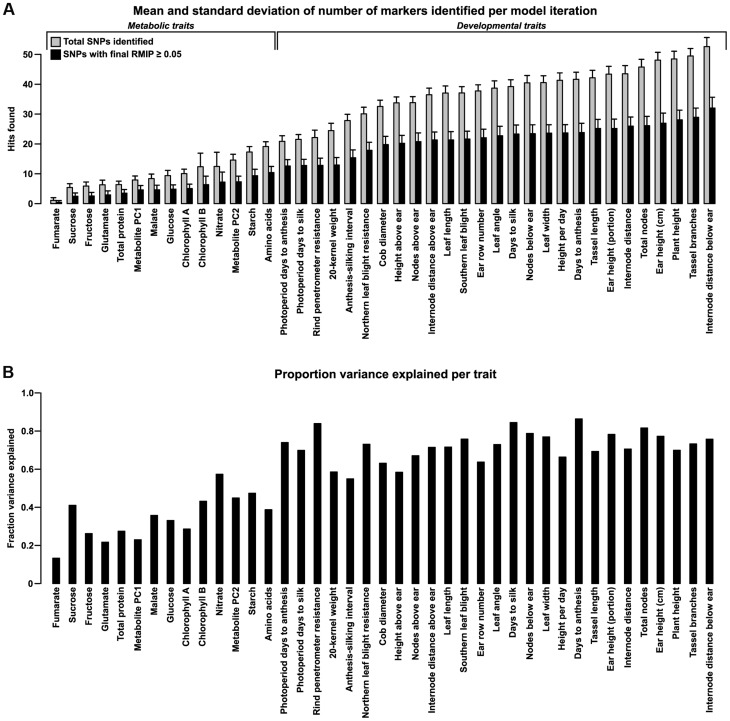
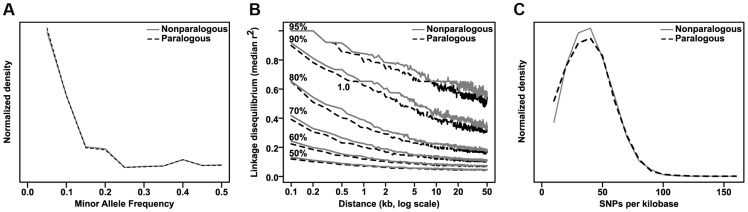
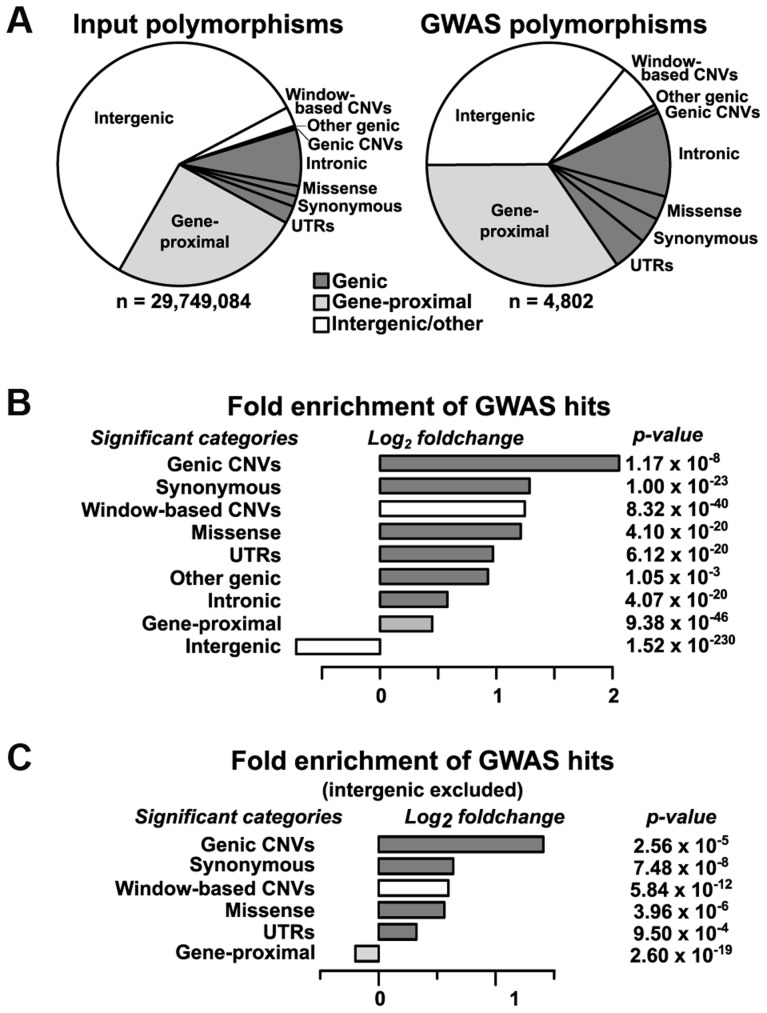
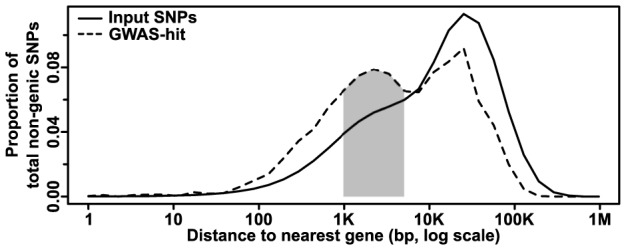
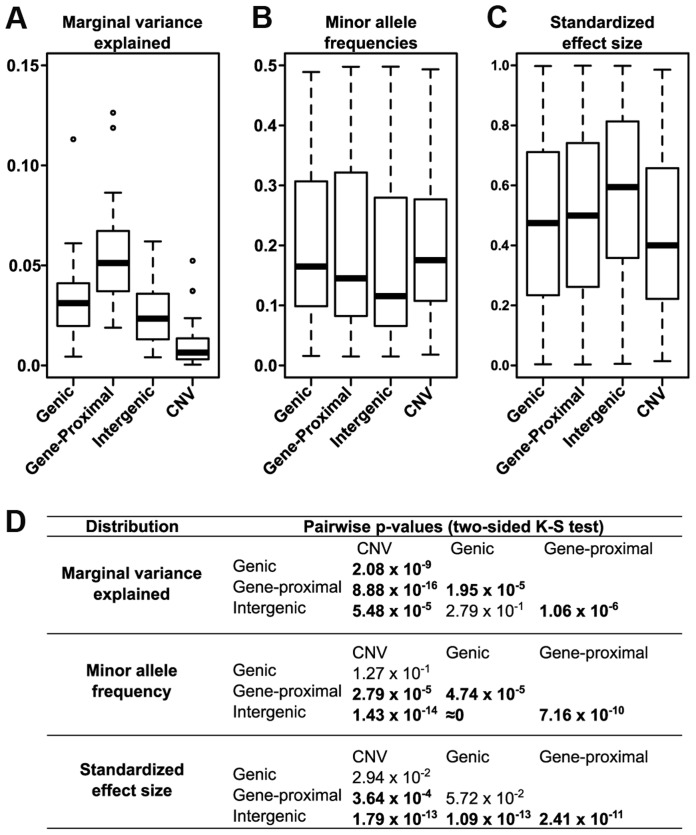
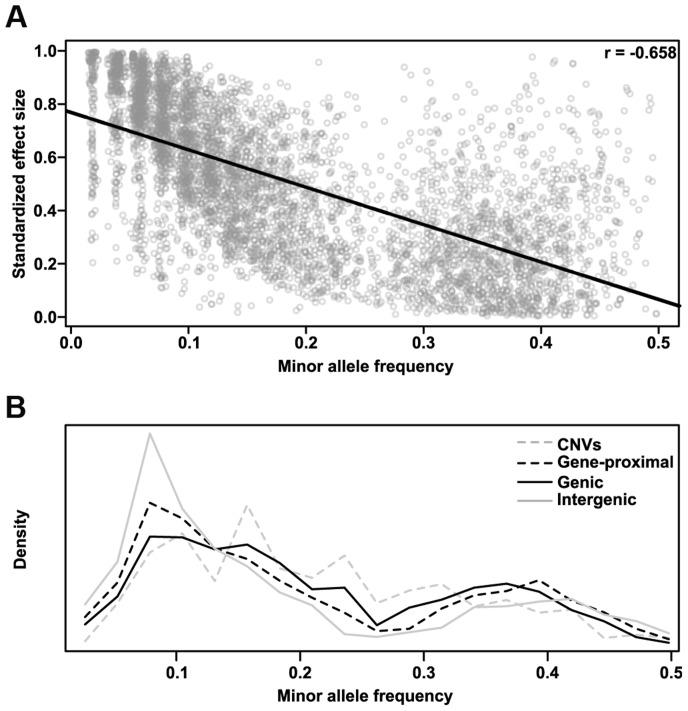
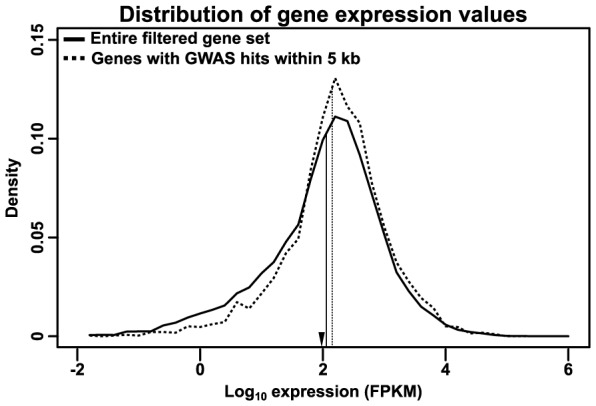
Phenotypic variation in natural populations results from a combination of genetic effects, environmental effects, and gene-by-environment interactions. Despite the vast amount of genomic data becoming available, many pressing questions remain about the nature of genetic mutations that underlie functional variation. We present the results of combining genome-wide association analysis of 41 different phenotypes in ∼ 5,000 inbred maize lines to analyze patterns of high-resolution genetic association among of 28.9 million single-nucleotide polymorphisms (SNPs) and ∼ 800,000 copy-number variants (CNVs). We show that genic and intergenic regions have opposite patterns of enrichment, minor allele frequencies, and effect sizes, implying tradeoffs among the probability that a given polymorphism will have an effect, the detectable size of that effect, and its frequency in the population. We also find that genes tagged by GWAS are enriched for regulatory functions and are ∼ 50% more likely to have a paralog than expected by chance, indicating that gene regulation and gene duplication are strong drivers of phenotypic variation. These results will likely apply to many other organisms, especially ones with large and complex genomes like maize.
自然种群中的表型变异是由遗传效应、环境效应以及基因与环境的相互作用共同作用的结果。尽管有大量的基因组数据可供使用,但关于构成功能变异基础的基因突变的本质,仍存在许多紧迫的问题。我们展示了对约5000个自交玉米品系中的41种不同表型进行全基因组关联分析的结果,以分析2890万个单核苷酸多态性(SNP)和约80万个拷贝数变异(CNV)之间的高分辨率遗传关联模式。我们发现基因区域和基因间区域在富集模式、次要等位基因频率和效应大小方面具有相反的模式,这意味着在给定多态性产生效应的概率、该效应的可检测大小及其在种群中的频率之间存在权衡。我们还发现,通过全基因组关联研究(GWAS)标记的基因在调控功能方面富集,并且具有旁系同源基因的可能性比随机预期的高约50%,这表明基因调控和基因复制是表型变异的强大驱动力。这些结果可能适用于许多其他生物,尤其是像玉米这样具有庞大而复杂基因组的生物。