Forsythe Stephen J, Dickins Benjamin, Jolley Keith A
School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK.
BMC Genomics. 2014 Dec 16;15(1):1121. doi: 10.1186/1471-2164-15-1121.
Following the association of Cronobacter spp. to several publicized fatal outbreaks in neonatal intensive care units of meningitis and necrotising enterocolitis, the World Health Organization (WHO) in 2004 requested the establishment of a molecular typing scheme to enable the international control of the organism. This paper presents the application of Next Generation Sequencing (NGS) to Cronobacter which has led to the establishment of the Cronobacter PubMLST genome and sequence definition database (http://pubmlst.org/cronobacter/) containing over 1000 isolates with metadata along with the recognition of specific clonal lineages linked to neonatal meningitis and adult infections
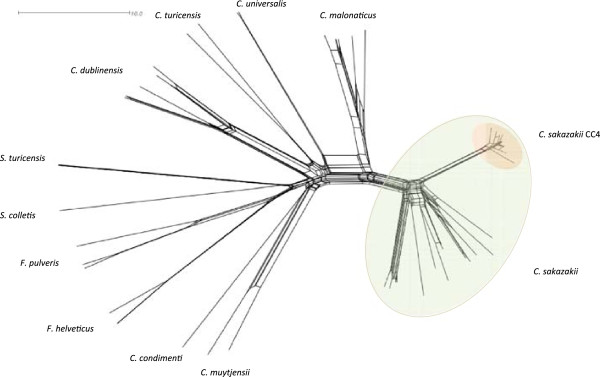
Whole genome sequencing and multilocus sequence typing (MLST) has supports the formal recognition of the genus Cronobacter composed of seven species to replace the former single species Enterobacter sakazakii. Applying the 7-loci MLST scheme to 1007 strains revealed 298 definable sequence types, yet only C. sakazakii clonal complex 4 (CC4) was principally associated with neonatal meningitis. This clonal lineage has been confirmed using ribosomal-MLST (51-loci) and whole genome-MLST (1865 loci) to analyse 107 whole genomes via the Cronobacter PubMLST database. This database has enabled the retrospective analysis of historic cases and outbreaks following re-identification of those strains.
The Cronobacter PubMLST database offers a central, open access, reliable sequence-based repository for researchers. It has the capacity to create new analysis schemes 'on the fly', and to integrate metadata (source, geographic distribution, clinical presentation). It is also expandable and adaptable to changes in taxonomy, and able to support the development of reliable detection methods of use to industry and regulatory authorities. Therefore it meets the WHO (2004) request for the establishment of a typing scheme for this emergent bacterial pathogen. Whole genome sequencing has additionally shown a range of potential virulence and environmental fitness traits which may account for the association of C. sakazakii CC4 pathogenicity, and propensity for neonatal CNS.
在阪崎肠杆菌与新生儿重症监护病房中几起广为人知的致命性脑膜炎和坏死性小肠结肠炎暴发事件相关联之后,世界卫生组织(WHO)于2004年要求建立一种分子分型方案,以实现对该微生物的国际管控。本文介绍了新一代测序(NGS)技术在阪崎肠杆菌上的应用,这导致了阪崎肠杆菌公共多位点序列分型(PubMLST)基因组和序列定义数据库(http://pubmlst.org/cronobacter/)的建立,该数据库包含1000多个带有元数据的分离株,并识别出与新生儿脑膜炎和成人感染相关的特定克隆谱系。
全基因组测序和多位点序列分型(MLST)支持正式认可由七个物种组成的阪崎肠杆菌属,以取代以前的单一物种阪崎肠杆菌。将7位点MLST方案应用于1007株菌株,揭示了298种可定义的序列类型,但只有阪崎肠杆菌克隆复合体4(CC4)主要与新生儿脑膜炎相关。通过阪崎肠杆菌PubMLST数据库,使用核糖体MLST(51位点)和全基因组MLST(1865位点)对107个全基因组进行分析,已证实了这种克隆谱系。该数据库能够在重新鉴定这些菌株后对历史病例和暴发事件进行回顾性分析。
阪崎肠杆菌PubMLST数据库为研究人员提供了一个集中的、开放获取的、基于序列的可靠储存库。它有能力“即时”创建新的分析方案,并整合元数据(来源、地理分布、临床表现)。它还具有可扩展性,能适应分类学的变化,并能够支持开发供行业和监管机构使用的可靠检测方法。因此,它满足了WHO(2004年)关于为这种新出现的细菌病原体建立分型方案的要求。全基因组测序还显示了一系列潜在的毒力和环境适应性特征,这可能解释了阪崎肠杆菌CC4的致病性以及对新生儿中枢神经系统的易感性。