Edwards Jeremy S, Atlas Susan R, Wilson Susan M, Cooper Candice F, Luo Li, Stidley Christine A
Molecular Genetics and Microbiology, and Chemical and Nuclear Engineering, 1 University of New Mexico, University of New Mexico Cancer Center, Albuquerque, NM 87131, USA.
Physics and Astronomy, Center for Advanced Research Computing, 1 University of New Mexico, University of New Mexico Cancer Center, Albuquerque, NM 87131, USA.
BMC Proc. 2014 Jun 17;8(Suppl 1 Genetic Analysis Workshop 18Vanessa Olmo):S104. doi: 10.1186/1753-6561-8-S1-S104. eCollection 2014.
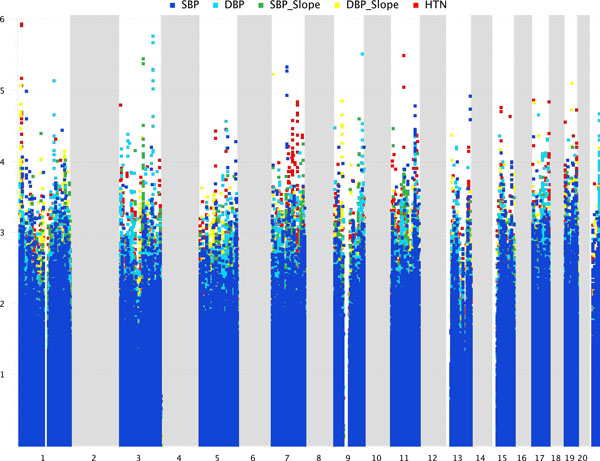
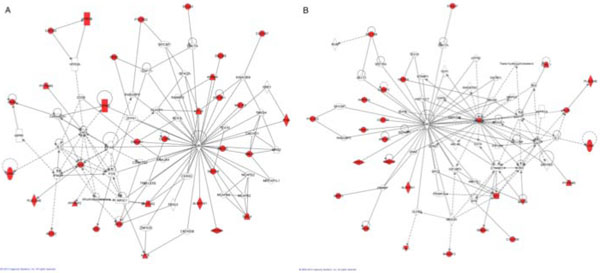
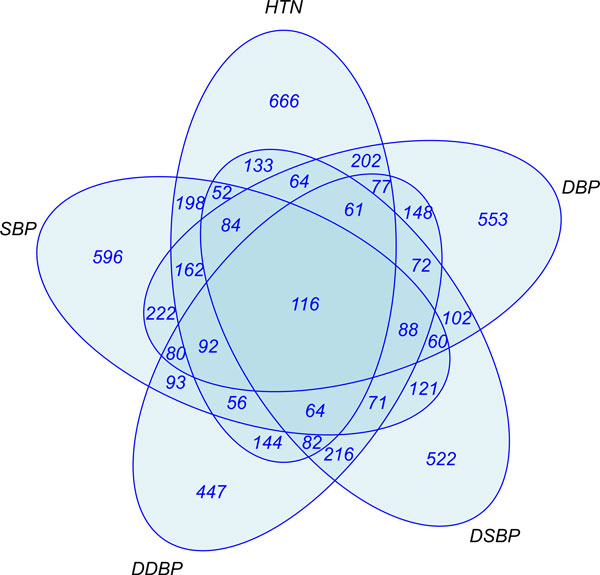
Genome wide association studies (GWAS) have been used to search for associations between genetic variants and a phenotypic trait of interest. New technologies, such as next-generation sequencing, hold the potential to revolutionize GWAS. However, millions of polymorphisms are identified with next-generation sequencing technology. Consequently, researchers must be careful when performing such a large number of statistical tests, and corrections are typically made to account for multiple testing. Additionally, for typical GWAS, the p value cutoff is set quite low (approximately <10(-8)). As a result of this p value stringency, it is likely that there are many true associations that do not meet this threshold. To account for this we have incorporated a priori biological knowledge to help identify true associations that may not have reached statistical significance. We propose the application of a pipelined series of statistical and bioinformatic methods, to enable the assessment of the association of genetic polymorphisms with a disease phenotype--here, hypertension--as well as the identification of statistically significant pathways of genes that may play a role in the disease process.
全基因组关联研究(GWAS)已被用于寻找基因变异与感兴趣的表型特征之间的关联。诸如新一代测序等新技术有潜力彻底改变GWAS。然而,新一代测序技术会识别出数百万个多态性。因此,研究人员在进行如此大量的统计测试时必须谨慎,并且通常会进行校正以考虑多重检验。此外,对于典型的GWAS,p值截止设定得相当低(约<10^(-8))。由于这种p值的严格性,很可能存在许多未达到此阈值的真实关联。为了解决这个问题,我们纳入了先验生物学知识,以帮助识别可能未达到统计学显著性的真实关联。我们建议应用一系列流水线式的统计和生物信息学方法,以评估基因多态性与疾病表型(此处为高血压)之间的关联,以及识别可能在疾病过程中起作用的具有统计学显著性的基因途径。