Perez Alberto, Yang Zheng, Bahar Ivet, Dill Ken A, MacCallum Justin L
Laufer Center for Physical and Quantitative Biology, Stony Brook University, Stony Brook, NY 11794-5252.
Department of Computational and Systems Biology, and Clinical & Translational Science Institute, School of Medicine, University of Pittsburgh, 3064 BST3, 3501 Fifth Ave, Pittsburgh, PA 15213.
J Chem Theory Comput. 2012 Oct 9;8(10):3985-3991. doi: 10.1021/ct300148f.
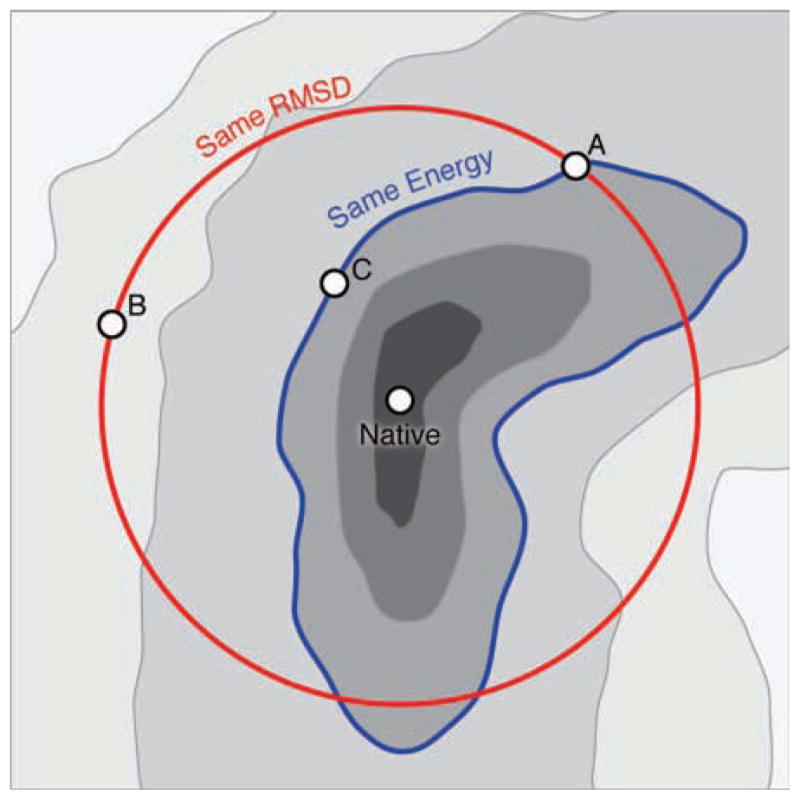
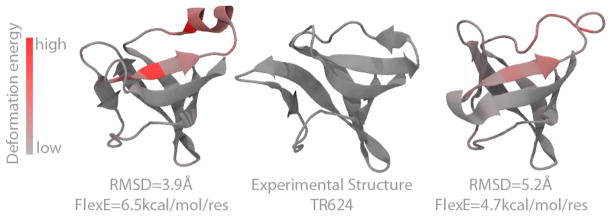
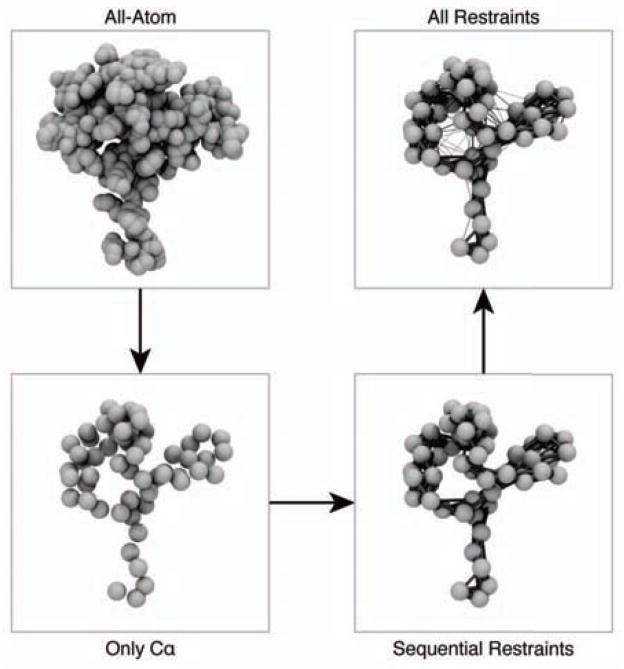
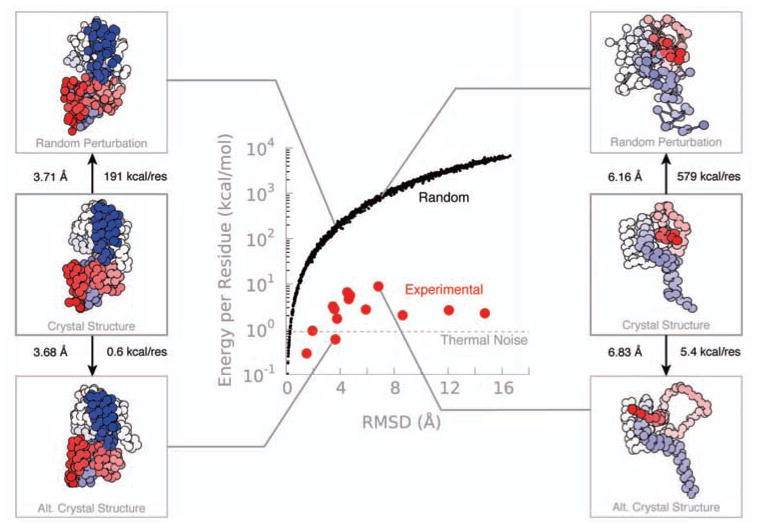
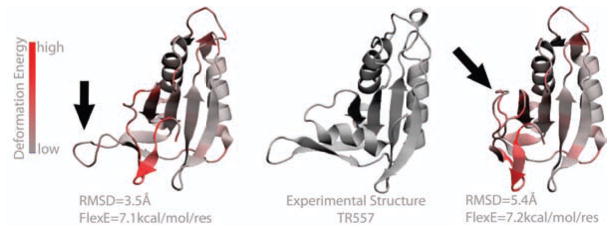
It is often valuable to compare protein structures to determine how similar they are. Structure comparison methods such as RMSD and GDT-TS are based solely on fixed geometry and do not take into account the intrinsic flexibility or energy landscape of the protein. We propose a method, which we call FlexE, that is based on a simple elastic network model and uses the deformation energy as measure of the similarity between two structures. FlexE can distinguish biologically relevant conformational changes from random changes, while existing geometry-based methods cannot. Additionally, FlexE incorporates the concept of thermal energy, which provides a rational way to determine when two models are "the same". FlexE provides a unique measure of the similarity between protein structures that is complementary to existing methods.
比较蛋白质结构以确定它们的相似程度通常很有价值。诸如均方根偏差(RMSD)和全局距离测试-全原子分数(GDT-TS)等结构比较方法仅基于固定的几何形状,并未考虑蛋白质的内在灵活性或能量态势。我们提出了一种方法,我们称之为FlexE,它基于一个简单的弹性网络模型,并使用变形能作为衡量两个结构之间相似性的指标。FlexE能够区分生物学上相关的构象变化和随机变化,而现有的基于几何形状的方法则无法做到。此外,FlexE纳入了热能的概念,这为确定两个模型何时“相同”提供了一种合理的方式。FlexE提供了一种独特的蛋白质结构相似性度量,它是对现有方法的补充。