Joshi Anagha
Division of Developmental Biology, The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Easter Bush Campus, Midlothian, EH25 8GR, UK.
BMC Bioinformatics. 2014 Dec 30;15(1):412. doi: 10.1186/s12859-014-0412-0.
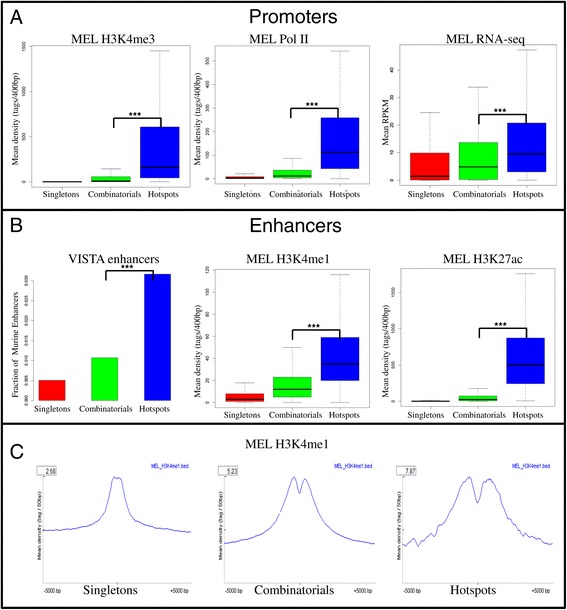
Transcriptional hotspots are defined as genomic regions bound by multiple factors. They have been identified recently as cell type specific enhancers regulating developmentally essential genes in many species such as worm, fly and humans. The in-depth analysis of hotspots across multiple cell types in same species still remains to be explored and can bring new biological insights.
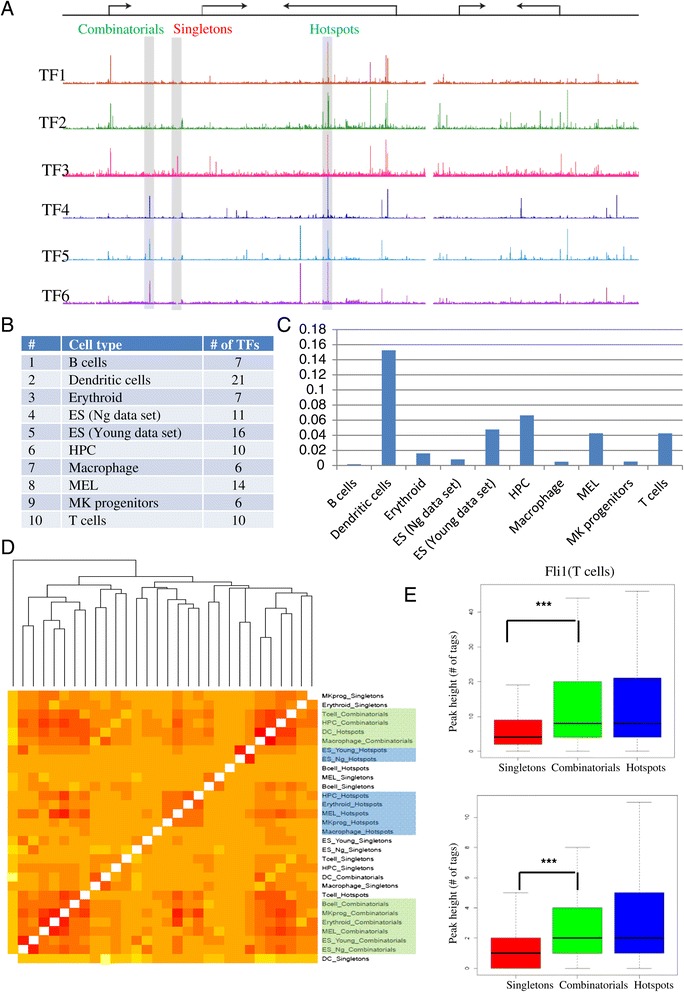
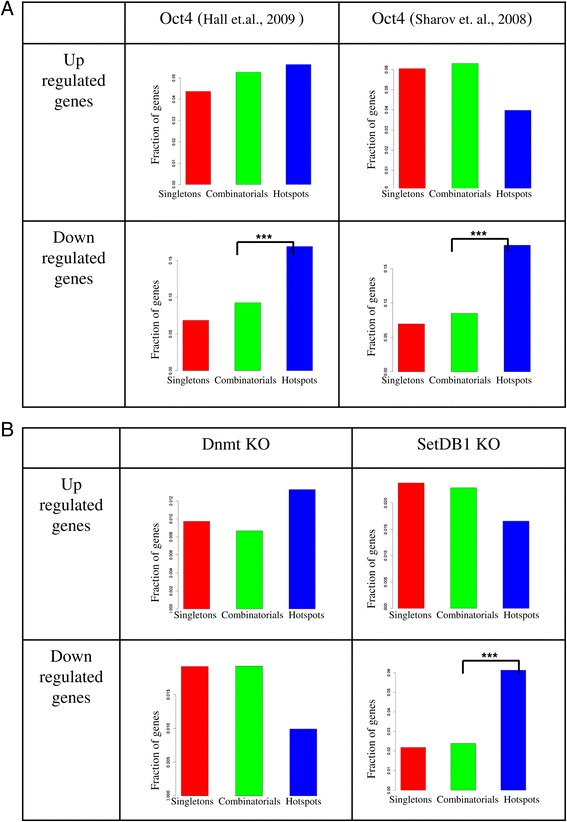
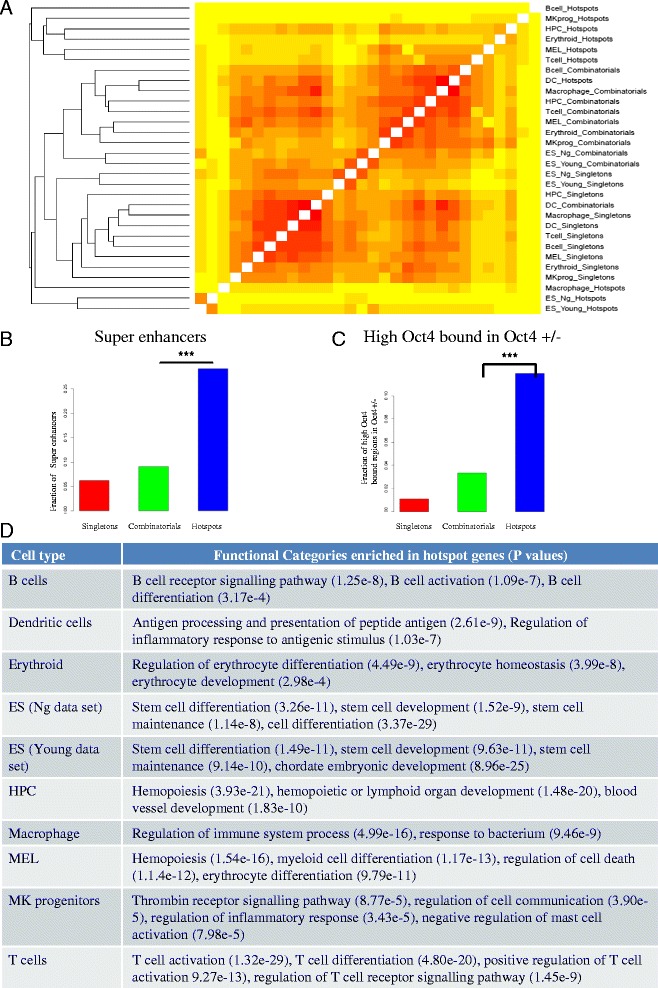
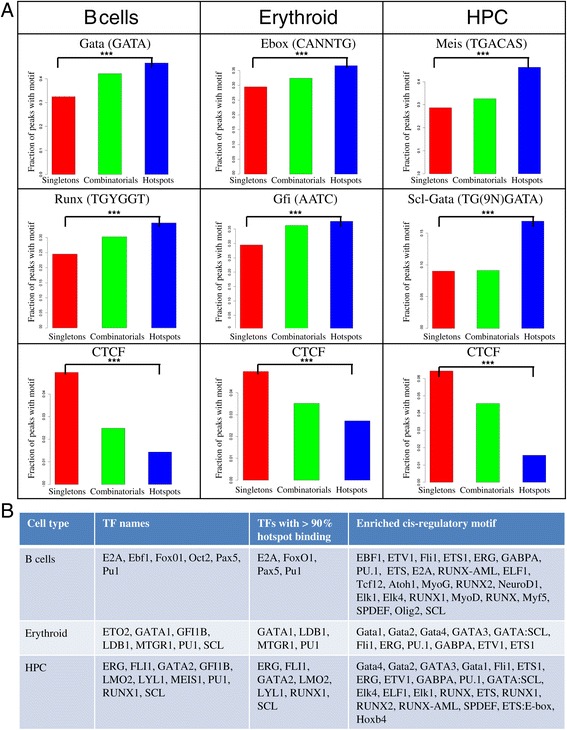
We therefore collected 108 transcription-related factor (TF) ChIP sequencing data sets in ten murine cell types and classified the peaks in each cell type in three groups according to binding occupancy as singletons (low-occupancy), combinatorials (mid-occupancy) and hotspots (high-occupancy). The peaks in the three groups clustered largely according to the occupancy, suggesting priming of genomic loci for mid occupancy irrespective of cell type. We then characterized hotspots for diverse structural functional properties. The genes neighbouring hotspots had a small overlap with hotspot genes in other cell types and were highly enriched for cell type specific function. Hotspots were enriched for sequence motifs of key TFs in that cell type and more than 90% of hotspots were occupied by pioneering factors. Though we did not find any sequence signature in the three groups, the H3K4me1 binding profile had bimodal peaks at hotspots, distinguishing hotspots from mono-modal H3K4me1 singletons. In ES cells, differentially expressed genes after perturbation of activators were enriched for hotspot genes suggesting hotspots primarily act as transcriptional activator hubs. Finally, we proposed that ES hotspots might be under control of SetDB1 and not DNMT for silencing.
Transcriptional hotspots are enriched for tissue specific enhancers near cell type specific highly expressed genes. In ES cells, they are predicted to act as transcriptional activator hubs and might be under SetDB1 control for silencing.
转录热点被定义为受多种因子结合的基因组区域。最近它们已被鉴定为细胞类型特异性增强子,在许多物种如线虫、果蝇和人类中调控发育必需基因。对同一物种多种细胞类型的热点进行深入分析仍有待探索,且可能带来新的生物学见解。
因此,我们收集了十种小鼠细胞类型中的108个转录相关因子(TF)染色质免疫沉淀测序数据集,并根据结合占有率将每种细胞类型中的峰分为三组:单峰(低占有率)、组合峰(中等占有率)和热点(高占有率)。这三组峰在很大程度上根据占有率聚类,表明无论细胞类型如何,基因组位点都为中等占有率做好了准备。然后我们对热点的各种结构功能特性进行了表征。与热点相邻的基因与其他细胞类型中的热点基因有少量重叠,并且在细胞类型特异性功能方面高度富集。热点在该细胞类型中富含关键转录因子的序列基序,超过90%的热点被先驱因子占据。虽然我们在这三组中未发现任何序列特征,但H3K4me1结合图谱在热点处有双峰,将热点与单峰H3K4me1区分开来。在胚胎干细胞中,激活剂扰动后差异表达的基因在热点基因中富集,表明热点主要作为转录激活中心发挥作用。最后,我们提出胚胎干细胞热点可能受SetDB1而非DNA甲基转移酶的控制进行沉默。
转录热点在细胞类型特异性高表达基因附近富含组织特异性增强子。在胚胎干细胞中,它们预计作为转录激活中心发挥作用,并且可能受SetDB1控制进行沉默。