Devlin Anna-Claire, Burr Karen, Borooah Shyamanga, Foster Joshua D, Cleary Elaine M, Geti Imbisaat, Vallier Ludovic, Shaw Christopher E, Chandran Siddharthan, Miles Gareth B
1] School of Psychology and Neuroscience, University of St. Andrews, Westburn Lane, St. Andrews KY16 9JP, UK [2] Euan MacDonald Centre for Motor Neurone Disease Research, Edinburgh EH16 4SB, UK.
1] Euan MacDonald Centre for Motor Neurone Disease Research, Edinburgh EH16 4SB, UK [2] Centre for Neuroregeneration and Medical Research Council Centre for Regenerative Medicine, University of Edinburgh, Edinburgh EH16 4UU, UK.
Nat Commun. 2015 Jan 12;6:5999. doi: 10.1038/ncomms6999.
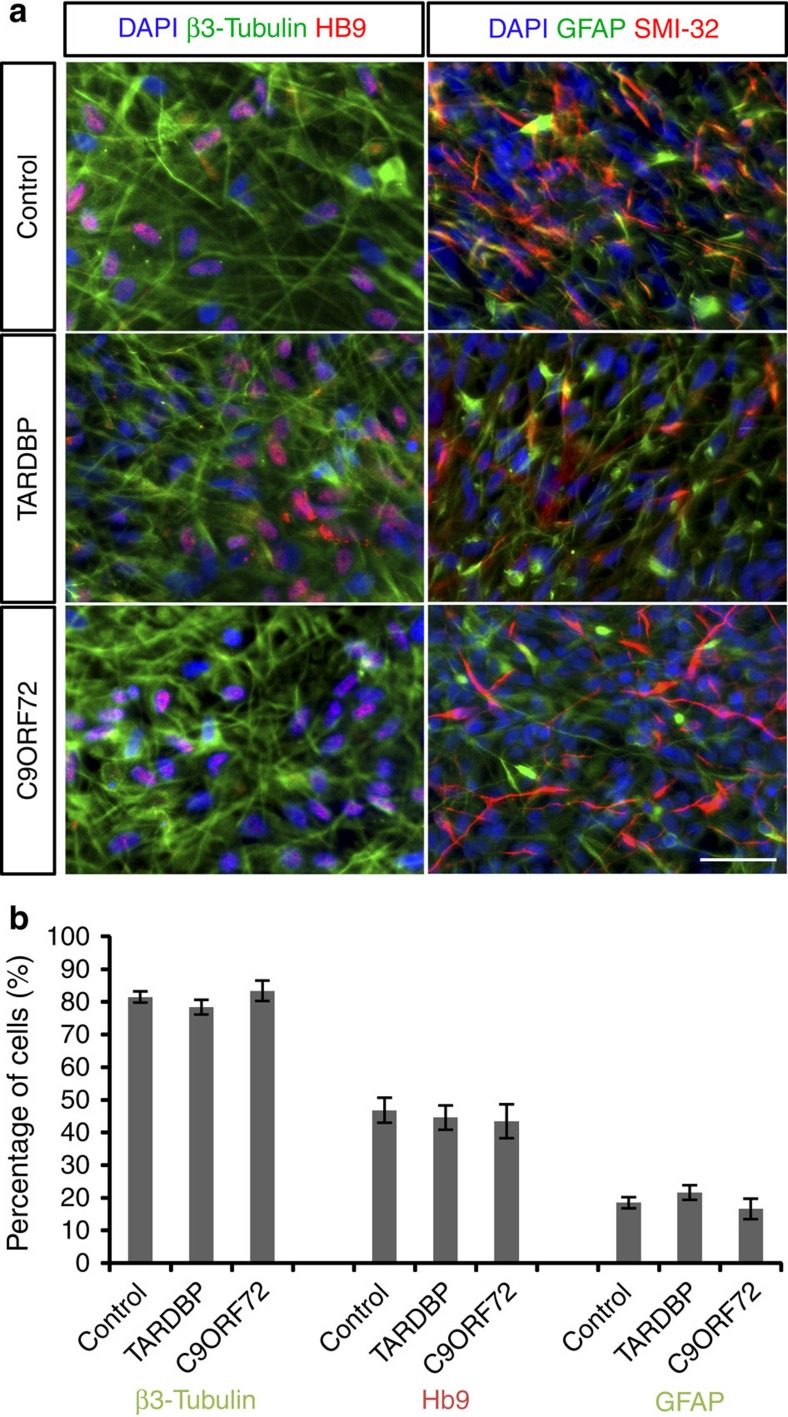
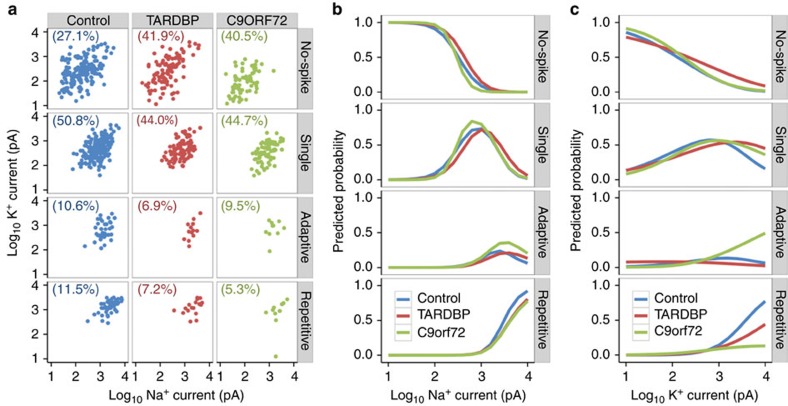
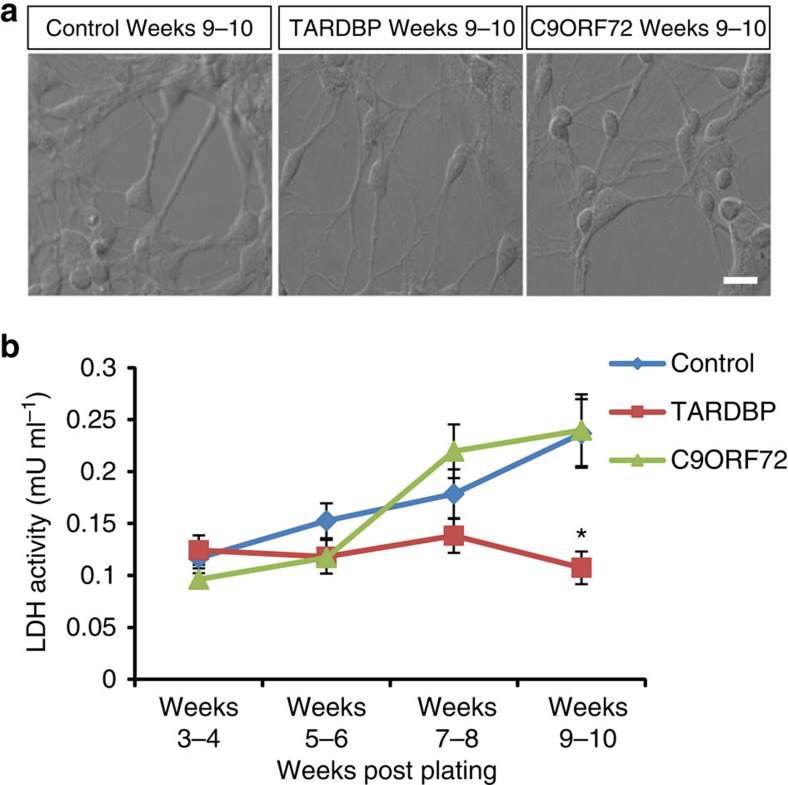
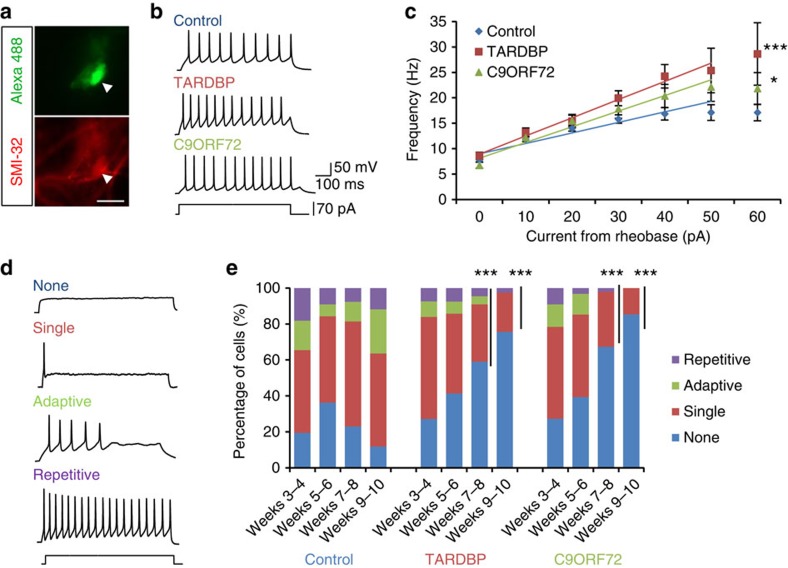
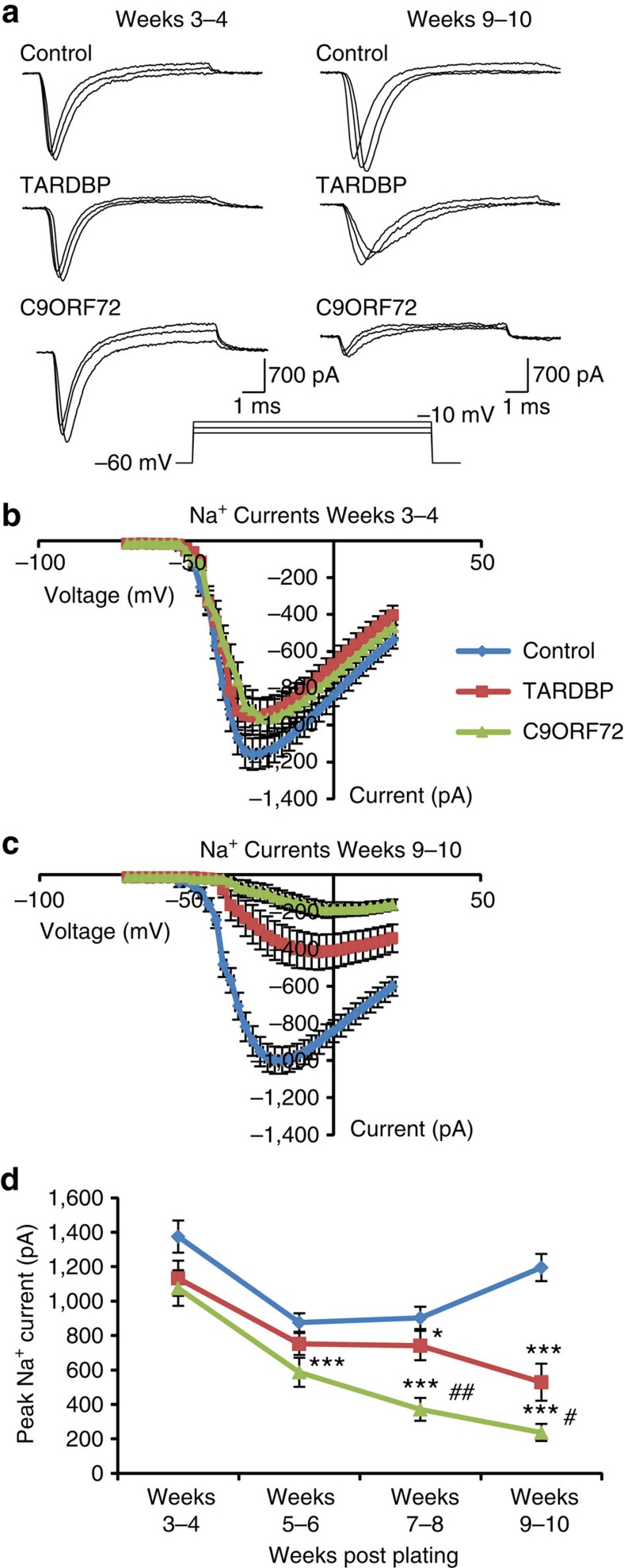
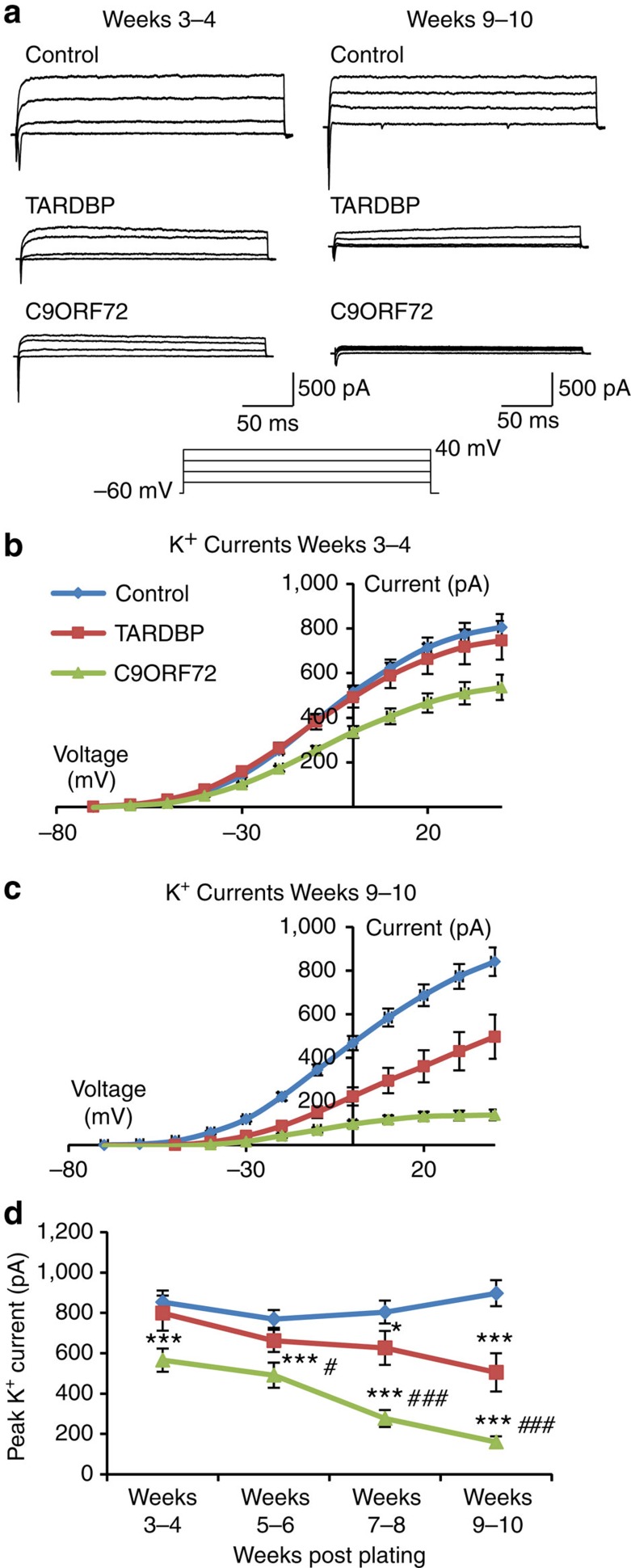
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease for which a greater understanding of early disease mechanisms is needed to reveal novel therapeutic targets. We report the use of human induced pluripotent stem cell (iPSC)-derived motoneurons (MNs) to study the pathophysiology of ALS. We demonstrate that MNs derived from iPSCs obtained from healthy individuals or patients harbouring TARDBP or C9ORF72 ALS-causing mutations are able to develop appropriate physiological properties. However, patient iPSC-derived MNs, independent of genotype, display an initial hyperexcitability followed by progressive loss of action potential output and synaptic activity. This loss of functional output reflects a progressive decrease in voltage-activated Na(+) and K(+) currents, which occurs in the absence of overt changes in cell viability. These data implicate early dysfunction or loss of ion channels as a convergent point that may contribute to the initiation of downstream degenerative pathways that ultimately lead to MN loss in ALS.
肌萎缩侧索硬化症(ALS)是一种毁灭性的神经退行性疾病,需要更深入地了解其早期疾病机制以揭示新的治疗靶点。我们报告了使用人类诱导多能干细胞(iPSC)衍生的运动神经元(MNs)来研究ALS的病理生理学。我们证明,从健康个体或携带TARDBP或C9ORF72 ALS致病突变的患者获得的iPSC衍生的MNs能够发展出适当的生理特性。然而,患者iPSC衍生的MNs,无论基因型如何,最初都表现出过度兴奋,随后动作电位输出和突触活动逐渐丧失。这种功能输出的丧失反映了电压激活的Na(+)和K(+)电流的逐渐减少,这在细胞活力没有明显变化的情况下发生。这些数据表明,离子通道的早期功能障碍或丧失是一个汇聚点,可能有助于启动下游退行性途径,最终导致ALS中MNs的丧失。