Jiang Yuchao, Oldridge Derek A, Diskin Sharon J, Zhang Nancy R
Genomics and Computational Biology Graduate Program, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Medical Scientist Training Program, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA Division of Oncology and Center for Childhood Cancer Research, The Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Nucleic Acids Res. 2015 Mar 31;43(6):e39. doi: 10.1093/nar/gku1363. Epub 2015 Jan 23.
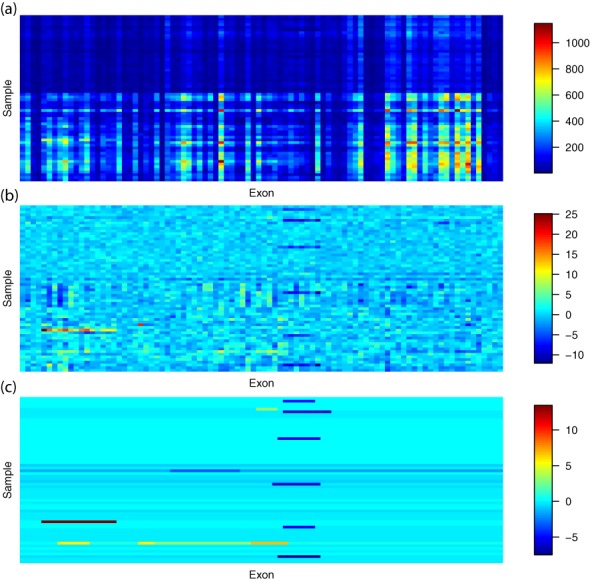
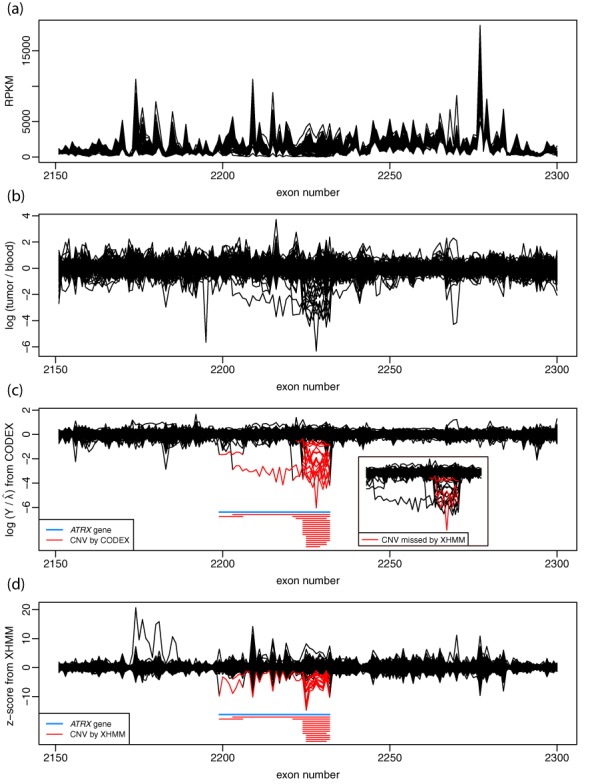
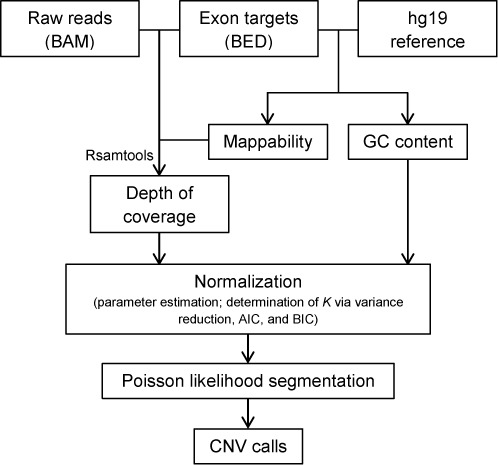
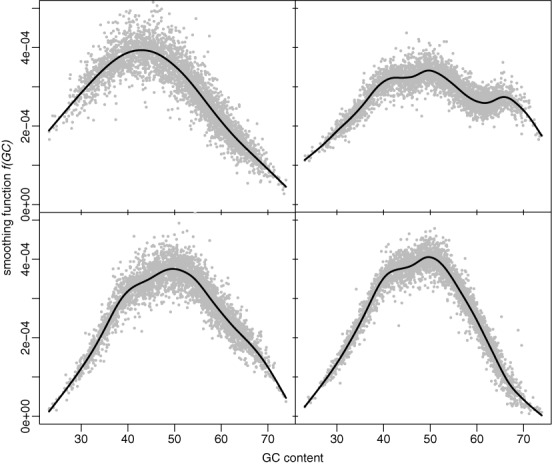
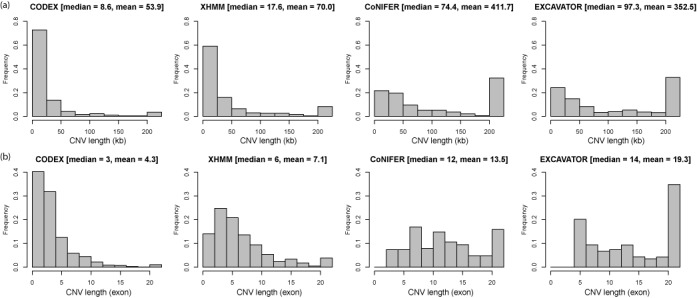
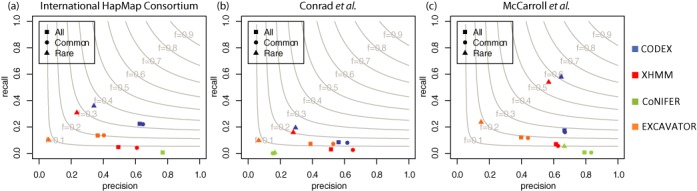
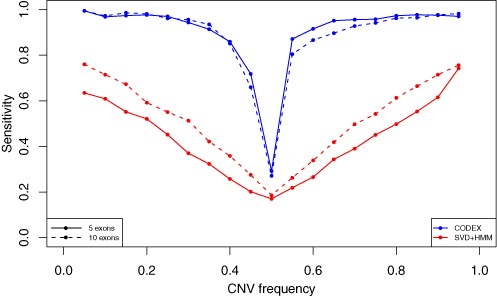
High-throughput sequencing of DNA coding regions has become a common way of assaying genomic variation in the study of human diseases. Copy number variation (CNV) is an important type of genomic variation, but detecting and characterizing CNV from exome sequencing is challenging due to the high level of biases and artifacts. We propose CODEX, a normalization and CNV calling procedure for whole exome sequencing data. The Poisson latent factor model in CODEX includes terms that specifically remove biases due to GC content, exon capture and amplification efficiency, and latent systemic artifacts. CODEX also includes a Poisson likelihood-based recursive segmentation procedure that explicitly models the count-based exome sequencing data. CODEX is compared to existing methods on a population analysis of HapMap samples from the 1000 Genomes Project, and shown to be more accurate on three microarray-based validation data sets. We further evaluate performance on 222 neuroblastoma samples with matched normals and focus on a well-studied rare somatic CNV within the ATRX gene. We show that the cross-sample normalization procedure of CODEX removes more noise than normalizing the tumor against the matched normal and that the segmentation procedure performs well in detecting CNVs with nested structures.
DNA编码区的高通量测序已成为人类疾病研究中检测基因组变异的常用方法。拷贝数变异(CNV)是一种重要的基因组变异类型,但由于偏差和伪影水平较高,从外显子组测序中检测和表征CNV具有挑战性。我们提出了CODEX,一种用于全外显子组测序数据的标准化和CNV检测程序。CODEX中的泊松潜在因子模型包含专门消除由于GC含量、外显子捕获和扩增效率以及潜在系统伪影导致的偏差的项。CODEX还包括一个基于泊松似然的递归分割程序,该程序明确地对基于计数的外显子组测序数据进行建模。在对来自千人基因组计划的HapMap样本进行群体分析时,将CODEX与现有方法进行了比较,结果表明在三个基于微阵列的验证数据集上,CODEX更为准确。我们进一步评估了在222个有匹配正常样本的神经母细胞瘤样本上的性能,并重点研究了ATRX基因内一个经过充分研究的罕见体细胞CNV。我们表明,CODEX的跨样本标准化程序比将肿瘤与匹配的正常样本进行标准化能去除更多噪声,并且分割程序在检测具有嵌套结构的CNV方面表现良好。