Gumienny Rafal, Zavolan Mihaela
Biozentrum, University of Basel and Swiss Institute of Bioinformatics, Klingelbergstrasse 50-70, 4056 Basel, Switzerland.
Biozentrum, University of Basel and Swiss Institute of Bioinformatics, Klingelbergstrasse 50-70, 4056 Basel, Switzerland
Nucleic Acids Res. 2015 Feb 18;43(3):1380-91. doi: 10.1093/nar/gkv050. Epub 2015 Jan 27.
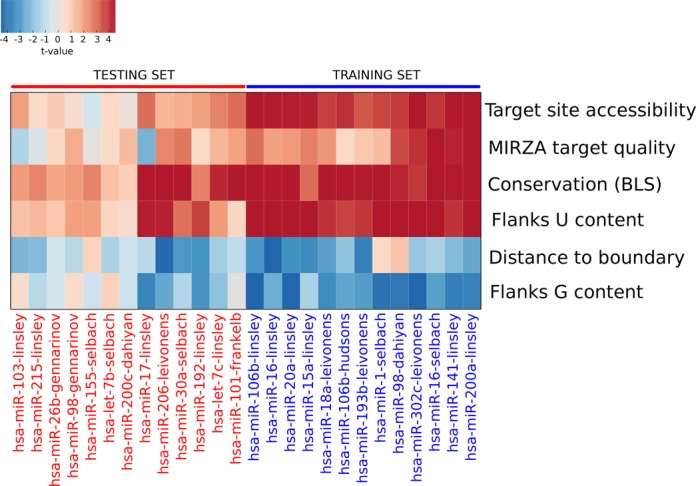
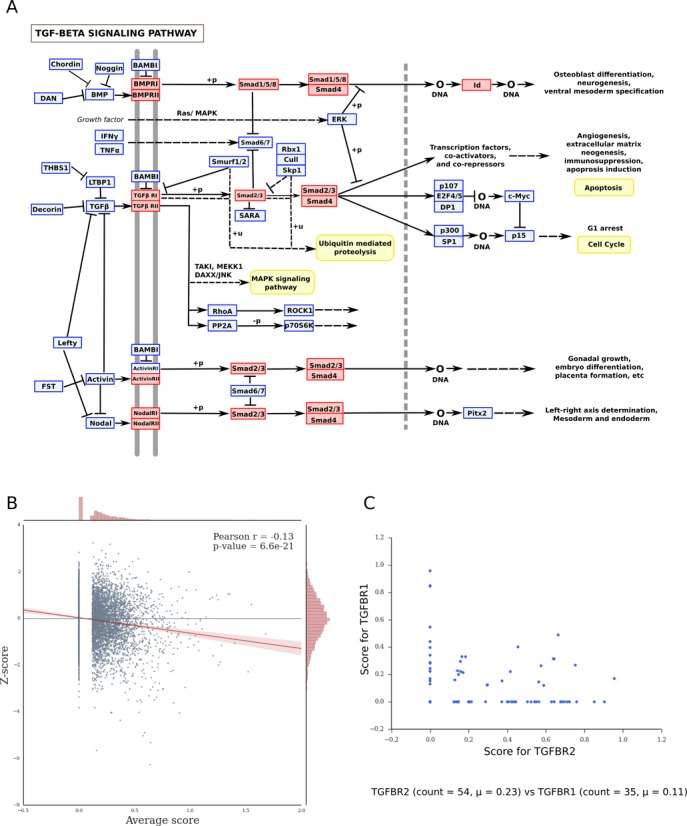
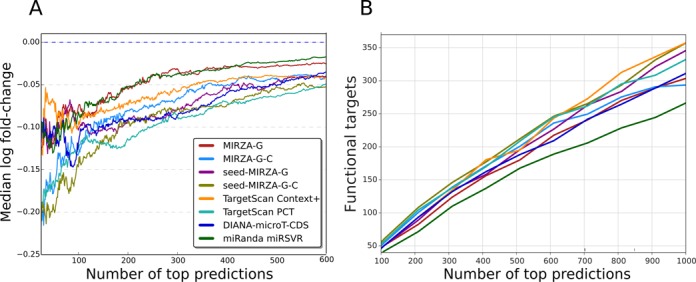
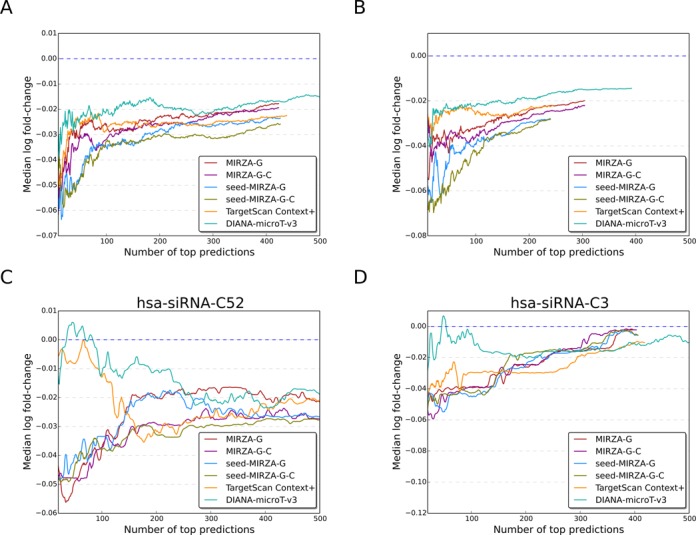
Small interfering RNA (siRNA)-mediated knock-down is a widely used experimental approach to characterizing gene function. Although siRNAs are designed to guide the cleavage of perfectly complementary mRNA targets, acting similarly to microRNAs (miRNAs), siRNAs down-regulate the expression of hundreds of genes to which they have only partial complementarity. Prediction of these siRNA 'off-targets' remains difficult, due to the incomplete understanding of siRNA/miRNA-target interactions. Combining a biophysical model of miRNA-target interaction with structure and sequence features of putative target sites we developed a suite of algorithms, MIRZA-G, for the prediction of miRNA targets and siRNA off-targets on a genome-wide scale. The MIRZA-G variant that uses evolutionary conservation performs better than currently available methods in predicting canonical miRNA target sites and in addition, it predicts non-canonical miRNA target sites with similarly high accuracy. Furthermore, MIRZA-G variants predict siRNA off-target sites with an accuracy unmatched by currently available programs. Thus, MIRZA-G may prove instrumental in the analysis of data resulting from large-scale siRNA screens.
小干扰RNA(siRNA)介导的基因敲低是一种广泛用于表征基因功能的实验方法。尽管siRNA被设计用于引导完全互补的mRNA靶标的切割,其作用类似于微小RNA(miRNA),但siRNA会下调数百个与其仅有部分互补性的基因的表达。由于对siRNA/miRNA-靶标相互作用的理解不完整,预测这些siRNA的“脱靶”仍然很困难。我们将miRNA-靶标相互作用的生物物理模型与假定靶位点的结构和序列特征相结合,开发了一套算法MIRZA-G,用于在全基因组范围内预测miRNA靶标和siRNA脱靶。在预测经典miRNA靶位点方面,使用进化保守性的MIRZA-G变体比目前可用的方法表现更好,此外,它以同样高的准确性预测非经典miRNA靶位点。此外,MIRZA-G变体预测siRNA脱靶位点的准确性是目前可用程序无法比拟的。因此,MIRZA-G可能在大规模siRNA筛选产生的数据的分析中发挥重要作用。