de Luna Noemí, Brull Astrid, Guiu Josep Maria, Lucia Alejandro, Martin Miguel Angel, Arenas Joaquin, Martí Ramon, Andreu Antoni L, Pinós Tomàs
Mitochondrial Pathology and Neuromuscular Disorders Laboratory, Vall d'Hebron Research Institute, Universitat Autònoma de Barcelona, Barcelona 08035, Spain Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid 28029, Spain.
Universidad Europea, Madrid 28670, Spain Instituto de Investigación 'i+12', Madrid 28041, Spain.
Dis Model Mech. 2015 May;8(5):467-72. doi: 10.1242/dmm.020230. Epub 2015 Mar 11.
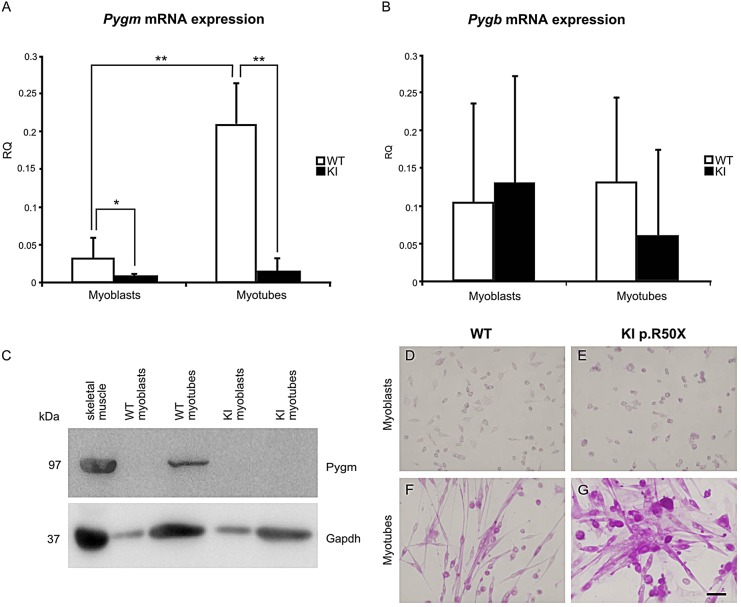
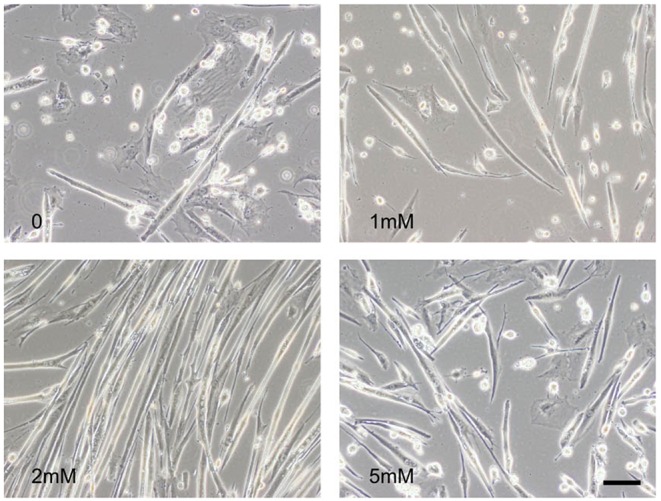
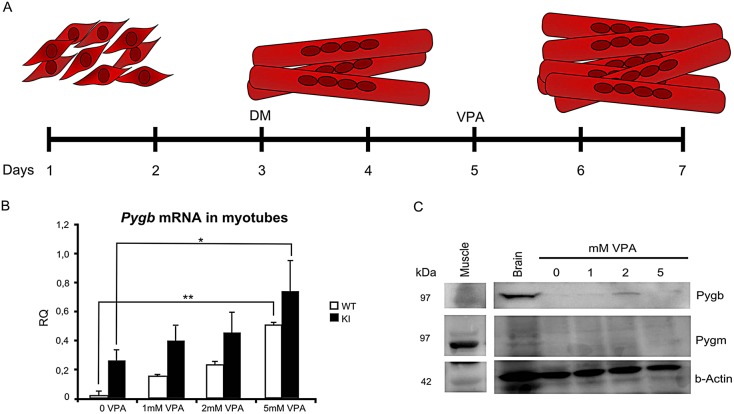
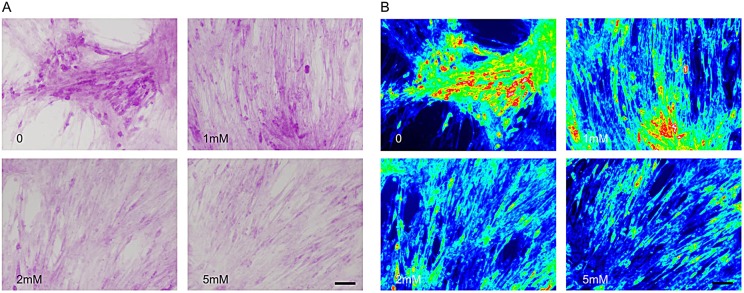
McArdle disease, also termed 'glycogen storage disease type V', is a disorder of skeletal muscle carbohydrate metabolism caused by inherited deficiency of the muscle-specific isoform of glycogen phosphorylase (GP-MM). It is an autosomic recessive disorder that is caused by mutations in the PYGM gene and typically presents with exercise intolerance, i.e. episodes of early exertional fatigue frequently accompanied by rhabdomyolysis and myoglobinuria. Muscle biopsies from affected individuals contain subsarcolemmal deposits of glycogen. Besides GP-MM, two other GP isoforms have been described: the liver (GP-LL) and brain (GP-BB) isoforms, which are encoded by the PYGL and PYGB genes, respectively; GP-BB is the main GP isoform found in human and rat foetal tissues, including the muscle, although its postnatal expression is dramatically reduced in the vast majority of differentiated tissues with the exception of brain and heart, where it remains as the major isoform. We developed a cell culture model from knock-in McArdle mice that mimics the glycogen accumulation and GP-MM deficiency observed in skeletal muscle from individuals with McArdle disease. We treated mouse primary skeletal muscle cultures in vitro with sodium valproate (VPA), a histone deacetylase inhibitor. After VPA treatment, myotubes expressed GP-BB and a dose-dependent decrease in glycogen accumulation was also observed. Thus, this in vitro model could be useful for high-throughput screening of new drugs to treat this disease. The immortalization of these primary skeletal muscle cultures could provide a never-ending source of cells for this experimental model. Furthermore, VPA could be considered as a gene-expression modulator, allowing compensatory expression of GP-BB and decreased glycogen accumulation in skeletal muscle of individuals with McArdle disease.
麦克尔迪氏病,也称为“糖原贮积病Ⅴ型”,是一种骨骼肌碳水化合物代谢紊乱疾病,由肌肉特异性糖原磷酸化酶同工酶(GP-MM)遗传性缺乏引起。它是一种常染色体隐性疾病,由PYGM基因突变所致,通常表现为运动不耐受,即早期运动性疲劳发作,常伴有横纹肌溶解和肌红蛋白尿。患病个体的肌肉活检显示糖原在肌膜下沉积。除了GP-MM,还描述了另外两种糖原磷酸化酶同工酶:肝脏(GP-LL)和脑(GP-BB)同工酶,它们分别由PYGL和PYGB基因编码;GP-BB是在人和大鼠胎儿组织(包括肌肉)中发现的主要糖原磷酸化酶同工酶,尽管其出生后的表达在绝大多数分化组织中显著降低,但在脑和心脏除外,在这些组织中它仍然是主要同工酶。我们利用敲入型麦克尔迪氏病小鼠建立了一种细胞培养模型,该模型模拟了麦克尔迪氏病患者骨骼肌中观察到的糖原积累和GP-MM缺乏情况。我们在体外用组蛋白脱乙酰酶抑制剂丙戊酸钠(VPA)处理小鼠原代骨骼肌培养物。VPA处理后,肌管表达GP-BB,并且还观察到糖原积累呈剂量依赖性减少。因此,这种体外模型可能有助于高通量筛选治疗该疾病的新药。这些原代骨骼肌培养物的永生化可为该实验模型提供源源不断的细胞来源。此外,VPA可被视为一种基因表达调节剂,它能使GP-BB代偿性表达,并减少麦克尔迪氏病患者骨骼肌中的糖原积累。