Ahlstrom Christina, Barkema Herman W, Stevenson Karen, Zadoks Ruth N, Biek Roman, Kao Rowland, Trewby Hannah, Haupstein Deb, Kelton David F, Fecteau Gilles, Labrecque Olivia, Keefe Greg P, McKenna Shawn L B, De Buck Jeroen
University of Calgary, Calgary, Alberta, Canada.
Moredun Research Institute, Penicuik, Scotland.
BMC Genomics. 2015 Mar 8;16(1):161. doi: 10.1186/s12864-015-1387-6.
Mycobacterium avium subsp. paratuberculosis (MAP), the causative bacterium of Johne's disease in dairy cattle, is widespread in the Canadian dairy industry and has significant economic and animal welfare implications. An understanding of the population dynamics of MAP can be used to identify introduction events, improve control efforts and target transmission pathways, although this requires an adequate understanding of MAP diversity and distribution between herds and across the country. Whole genome sequencing (WGS) offers a detailed assessment of the SNP-level diversity and genetic relationship of isolates, whereas several molecular typing techniques used to investigate the molecular epidemiology of MAP, such as variable number of tandem repeat (VNTR) typing, target relatively unstable repetitive elements in the genome that may be too unpredictable to draw accurate conclusions. The objective of this study was to evaluate the diversity of bovine MAP isolates in Canadian dairy herds using WGS and then determine if VNTR typing can distinguish truly related and unrelated isolates.
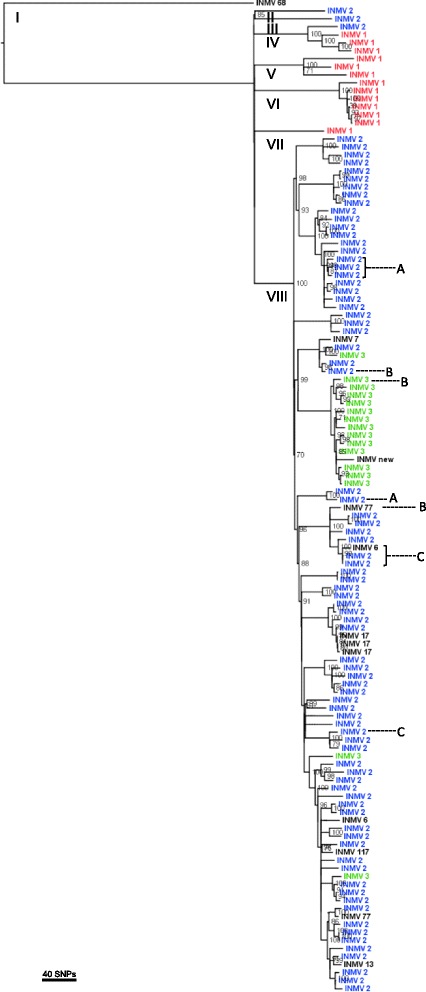
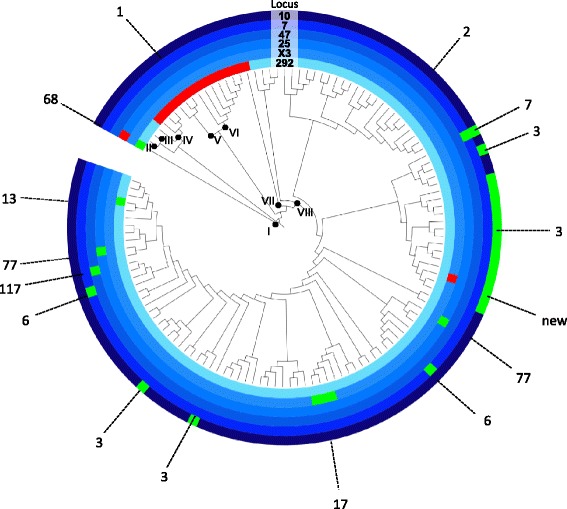
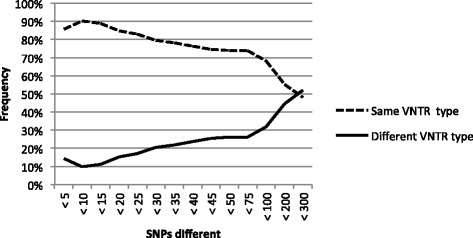
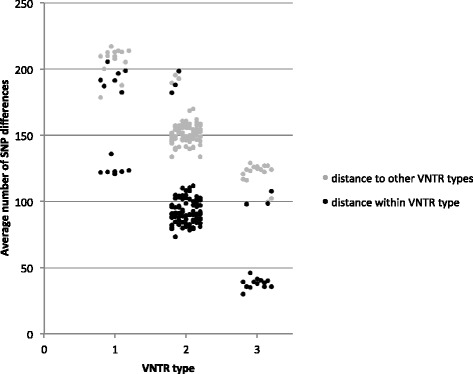
Phylogenetic analysis based on 3,039 SNPs identified through WGS of 124 MAP isolates identified eight genetically distinct subtypes in dairy herds from seven Canadian provinces, with the dominant type including over 80% of MAP isolates. VNTR typing of 527 MAP isolates identified 12 types, including "bison type" isolates, from seven different herds. At a national level, MAP isolates differed from each other by 1-2 to 239-240 SNPs, regardless of whether they belonged to the same or different VNTR types. A herd-level analysis of MAP isolates demonstrated that VNTR typing may both over-estimate and under-estimate the relatedness of MAP isolates found within a single herd.
The presence of multiple MAP subtypes in Canada suggests multiple introductions into the country including what has now become one dominant type, an important finding for Johne's disease control. VNTR typing often failed to identify closely and distantly related isolates, limiting the applicability of using this typing scheme to study the molecular epidemiology of MAP at a national and herd-level.
副结核分枝杆菌(MAP)是奶牛约翰氏病的致病菌,在加拿大乳制品行业广泛存在,对经济和动物福利有重大影响。了解MAP的种群动态可用于识别引入事件、加强防控措施并确定传播途径,不过这需要充分了解MAP在畜群间以及全国范围内的多样性和分布情况。全基因组测序(WGS)可详细评估分离株的单核苷酸多态性(SNP)水平多样性和遗传关系,而用于研究MAP分子流行病学的几种分子分型技术,如可变数目串联重复序列(VNTR)分型,针对的是基因组中相对不稳定的重复元件,这些元件可能过于不可预测,无法得出准确结论。本研究的目的是利用WGS评估加拿大奶牛群中牛源MAP分离株的多样性,然后确定VNTR分型能否区分真正相关和不相关的分离株。
基于对124株MAP分离株进行WGS鉴定出的3039个SNP进行系统发育分析,在来自加拿大七个省份的奶牛群中确定了八个遗传上不同的亚型,其中占主导地位的亚型包含超过80%的MAP分离株。对527株MAP分离株进行VNTR分型,从七个不同畜群中鉴定出12种类型,包括“野牛型”分离株。在全国范围内,MAP分离株彼此之间相差1 - 2至239 - 240个SNP,无论它们属于相同还是不同的VNTR类型。对MAP分离株进行畜群水平分析表明,VNTR分型可能高估也可能低估单个畜群中MAP分离株的相关性。
加拿大存在多种MAP亚型,表明多次引入该国,包括现已成为一种主导类型的亚型,这是约翰氏病防控的一个重要发现。VNTR分型常常无法识别密切相关和远距离相关的分离株,限制了使用该分型方案在全国和畜群水平研究MAP分子流行病学的适用性。