Genheden Samuel, Ryde Ulf
University of Southampton, School of Chemistry , Highfield, SO17 1BJ, Southampton , UK.
Expert Opin Drug Discov. 2015 May;10(5):449-61. doi: 10.1517/17460441.2015.1032936. Epub 2015 Apr 2.
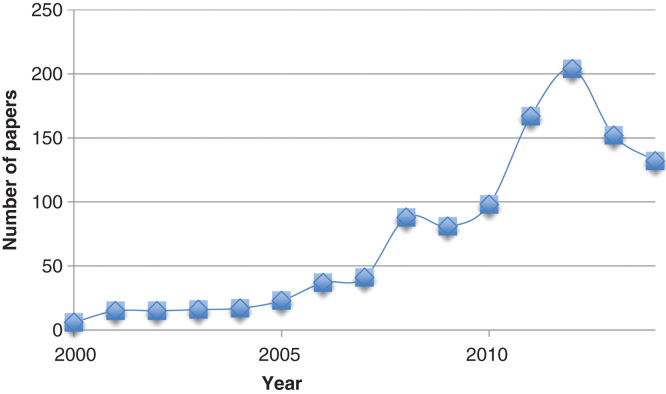
The molecular mechanics energies combined with the Poisson-Boltzmann or generalized Born and surface area continuum solvation (MM/PBSA and MM/GBSA) methods are popular approaches to estimate the free energy of the binding of small ligands to biological macromolecules. They are typically based on molecular dynamics simulations of the receptor-ligand complex and are therefore intermediate in both accuracy and computational effort between empirical scoring and strict alchemical perturbation methods. They have been applied to a large number of systems with varying success.
The authors review the use of MM/PBSA and MM/GBSA methods to calculate ligand-binding affinities, with an emphasis on calibration, testing and validation, as well as attempts to improve the methods, rather than on specific applications.
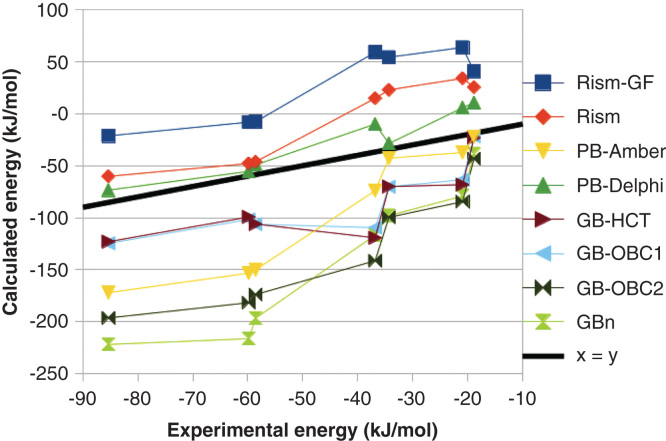
MM/PBSA and MM/GBSA are attractive approaches owing to their modular nature and that they do not require calculations on a training set. They have been used successfully to reproduce and rationalize experimental findings and to improve the results of virtual screening and docking. However, they contain several crude and questionable approximations, for example, the lack of conformational entropy and information about the number and free energy of water molecules in the binding site. Moreover, there are many variants of the method and their performance varies strongly with the tested system. Likewise, most attempts to ameliorate the methods with more accurate approaches, for example, quantum-mechanical calculations, polarizable force fields or improved solvation have deteriorated the results.
分子力学能量结合泊松-玻尔兹曼或广义玻恩及表面积连续介质溶剂化(MM/PBSA和MM/GBSA)方法是估算小分子配体与生物大分子结合自由能的常用方法。它们通常基于受体-配体复合物的分子动力学模拟,因此在准确性和计算量方面介于经验评分和严格的炼金术微扰方法之间。它们已被应用于大量系统,取得了不同程度的成功。
作者回顾了MM/PBSA和MM/GBSA方法在计算配体结合亲和力方面的应用,重点是校准、测试和验证,以及改进这些方法的尝试,而非具体应用。
MM/PBSA和MM/GBSA因其模块化性质且无需在训练集上进行计算而颇具吸引力。它们已成功用于重现实验结果并使其合理化,以及改进虚拟筛选和对接的结果。然而,它们包含一些粗糙且有问题的近似,例如缺乏构象熵以及结合位点水分子数量和自由能的信息。此外,该方法有许多变体,其性能会因测试系统的不同而有很大差异。同样,大多数用更精确方法(如量子力学计算、可极化力场或改进的溶剂化)改进这些方法的尝试都使结果变差。