Schramm Vern L
Department of Biochemistry, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, New York 10461, United States.
Acc Chem Res. 2015 Apr 21;48(4):1032-9. doi: 10.1021/acs.accounts.5b00002. Epub 2015 Apr 7.
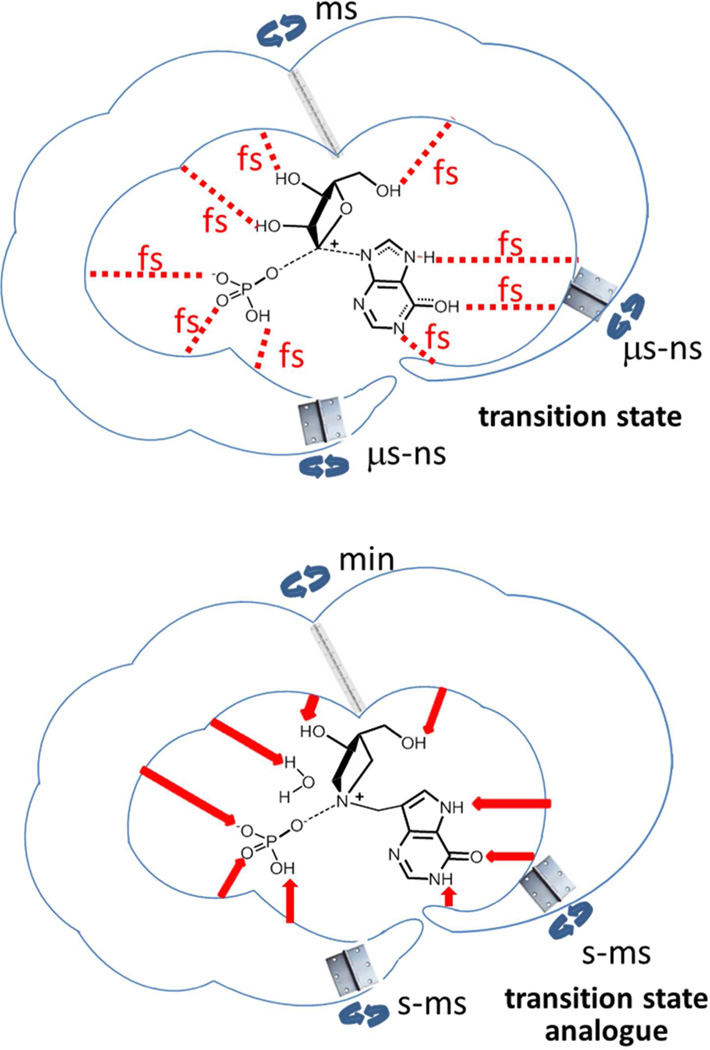
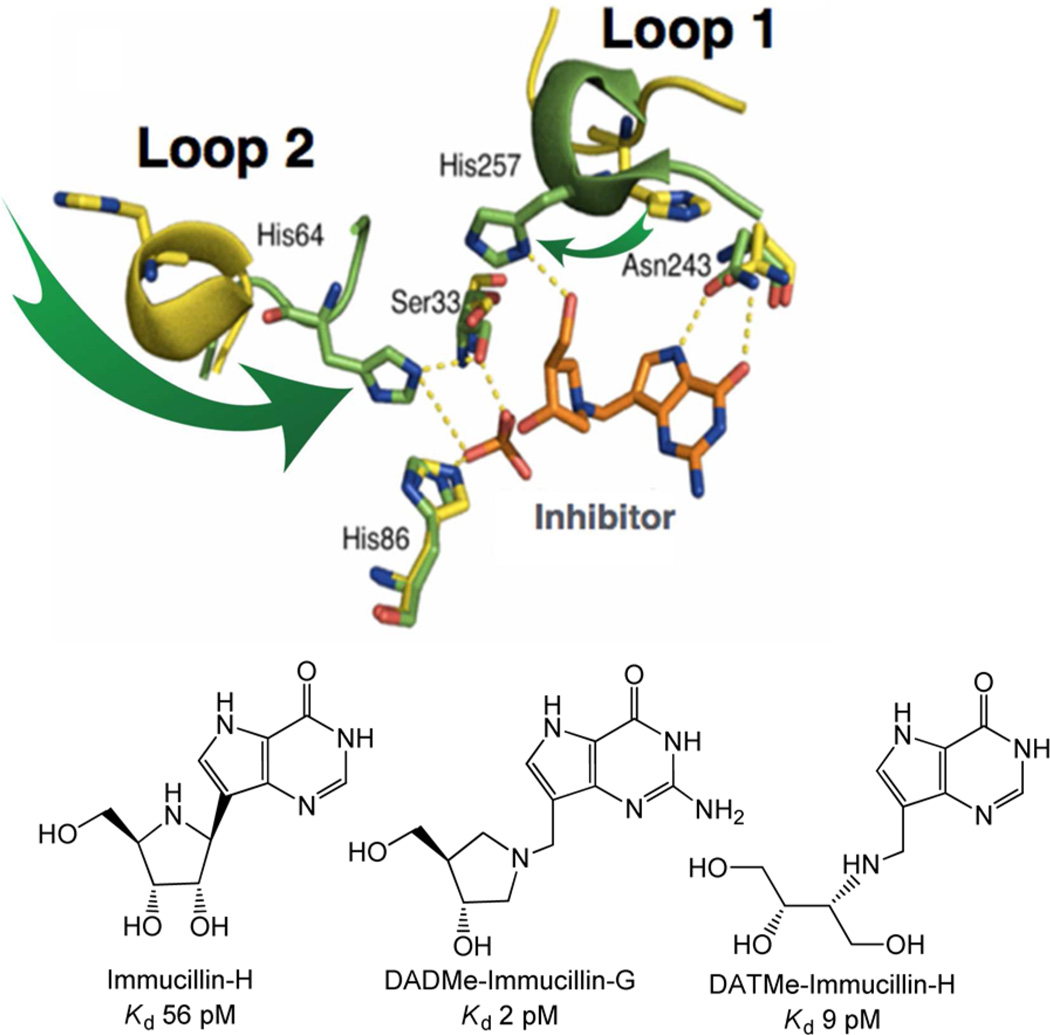
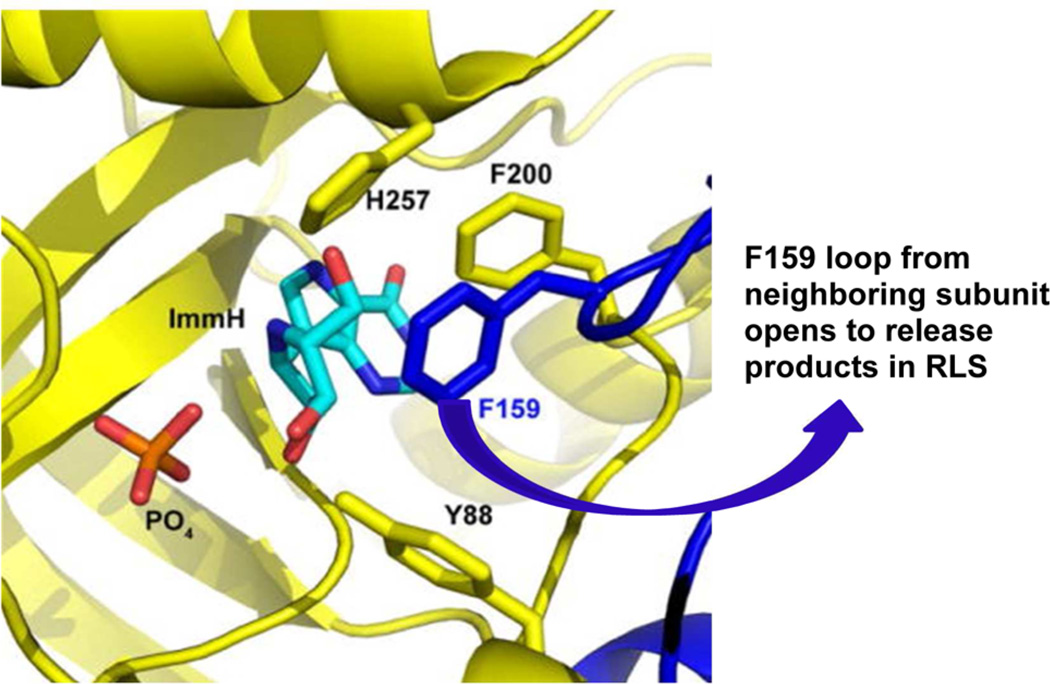
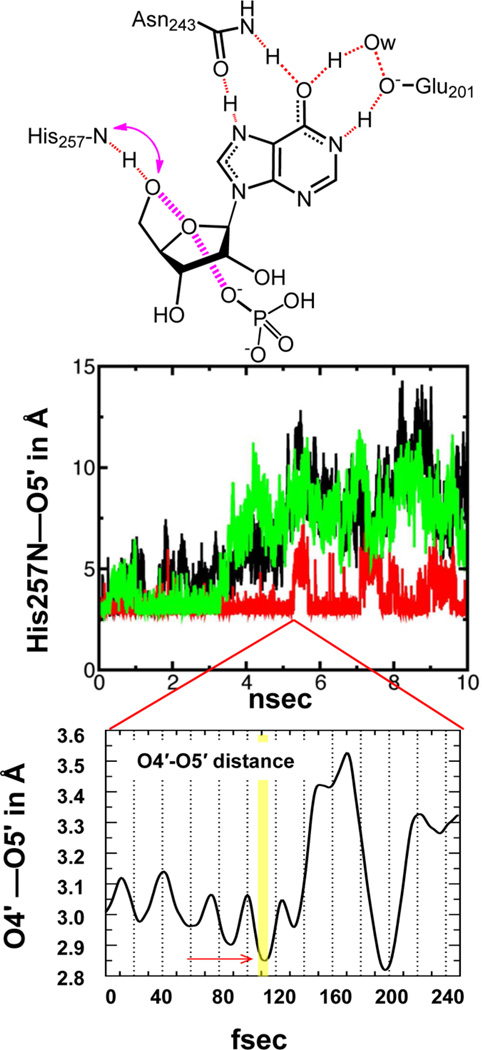
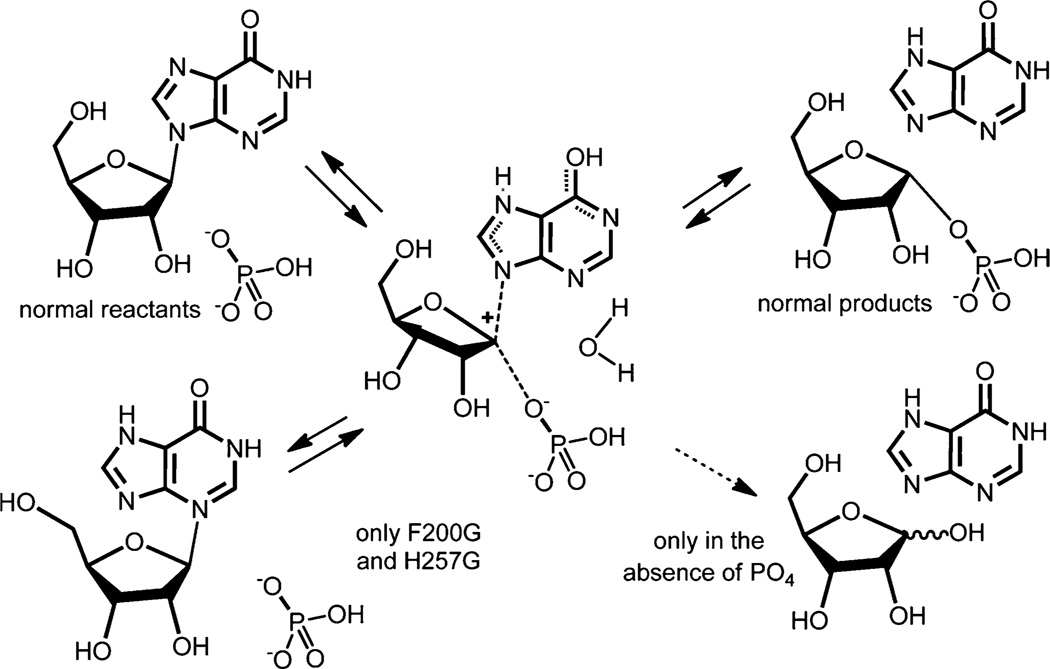
Enzymatic transition states have lifetimes of a few femtoseconds (fs). Computational analysis of enzyme motions leading to transition state formation suggests that local catalytic site motions on the fs time scale provide the mechanism to locate transition states. An experimental test of protein fs motion and its relation to transition state formation can be provided by isotopically heavy proteins. Heavy enzymes have predictable mass-altered bond vibration states without altered electrostatic properties, according to the Born-Oppenheimer approximation. On-enzyme chemistry is slowed in most heavy proteins, consistent with altered protein bond frequencies slowing the search for the transition state. In other heavy enzymes, structural changes involved in reactant binding and release are also influenced. Slow protein motions associated with substrate binding and catalytic site preorganization are essential to allow the subsequent fs motions to locate the transition state and to facilitate the efficient release of products. In the catalytically competent geometry, local groups move in stochastic atomic motion on the fs time scale, within transition state-accessible conformations created by slower protein motions. The fs time scale for the transition state motions does not permit thermodynamic equilibrium between the transition state and stable enzyme states. Isotopically heavy enzymes provide a diagnostic tool for fast coupled protein motions to transition state formation and mass-dependent conformational changes. The binding of transition state analogue inhibitors is the opposite in catalytic time scale to formation of the transition state but is related by similar geometries of the enzyme-transition state and enzyme-inhibitor interactions. While enzymatic transition states have lifetimes as short as 10(-15) s, transition state analogues can bind tightly to enzymes with release rates greater than 10(3) s. Tight-binding transition state analogues stabilize the rare but evolved enzymatic geometry to form the transition state. Evolution to efficient catalysis optimized this geometry and its stabilization by a transition state mimic results in tight binding. Release rates of transition state analogues are orders of magnitude slower than product release in normal catalytic function. During catalysis, product release is facilitated by altered chemistry. Compared to the weak associations found in Michaelis complexes, transition state analogues involve strong interactions related to those in the transition state. Optimum binding of transition state analogues occurs when the complex retains the system motions intrinsic to transition state formation. Conserved dynamic motion retains the entropic components of inhibitor complexes, improving the thermodynamics of analogue binding.
酶促过渡态的寿命为几飞秒(fs)。对导致过渡态形成的酶运动进行的计算分析表明,飞秒时间尺度上的局部催化位点运动提供了定位过渡态的机制。同位素标记的重蛋白质可对蛋白质的飞秒运动及其与过渡态形成的关系进行实验测试。根据玻恩 - 奥本海默近似,重酶具有可预测的质量改变的键振动状态,而静电性质不变。在大多数重蛋白质中,酶上的化学反应会减慢,这与蛋白质键频率的改变减缓了对过渡态的搜索一致。在其他重酶中,反应物结合和释放所涉及的结构变化也会受到影响。与底物结合和催化位点预组织相关的缓慢蛋白质运动对于使随后的飞秒运动能够定位过渡态并促进产物的有效释放至关重要。在具有催化活性的几何结构中,局部基团在飞秒时间尺度上以随机原子运动的方式移动,处于由较慢蛋白质运动产生的可接近过渡态的构象内。过渡态运动的飞秒时间尺度不允许过渡态与稳定酶状态之间达到热力学平衡。同位素标记的重酶为快速耦合蛋白质运动与过渡态形成以及质量依赖性构象变化提供了一种诊断工具。过渡态类似物抑制剂的结合在催化时间尺度上与过渡态的形成相反,但通过酶 - 过渡态和酶 - 抑制剂相互作用的相似几何结构相关联。虽然酶促过渡态的寿命短至10^(-15) 秒,但过渡态类似物可以以大于10^3 秒的释放速率紧密结合到酶上。紧密结合的过渡态类似物稳定了罕见但已进化的酶促几何结构以形成过渡态。向高效催化的进化优化了这种几何结构,并且通过过渡态模拟物对其进行稳定导致紧密结合。过渡态类似物的释放速率比正常催化功能中产物释放的速率慢几个数量级。在催化过程中,化学变化促进了产物释放。与米氏复合物中发现的弱结合相比,过渡态类似物涉及与过渡态中相关的强相互作用。当复合物保留过渡态形成所固有的系统运动时,过渡态类似物会发生最佳结合。保守的动态运动保留了抑制剂复合物的熵成分,改善了类似物结合的热力学。