Kaus Joseph W, McCammon J Andrew
†Department of Chemistry and Biochemistry,‡Center for Theoretical Biological Physics,¶Department of Pharmacology, and §Howard Hughes Medical Institute, University of California San Diego, La Jolla, California 92093-0365, United States.
J Phys Chem B. 2015 May 21;119(20):6190-7. doi: 10.1021/acs.jpcb.5b02348. Epub 2015 May 8.
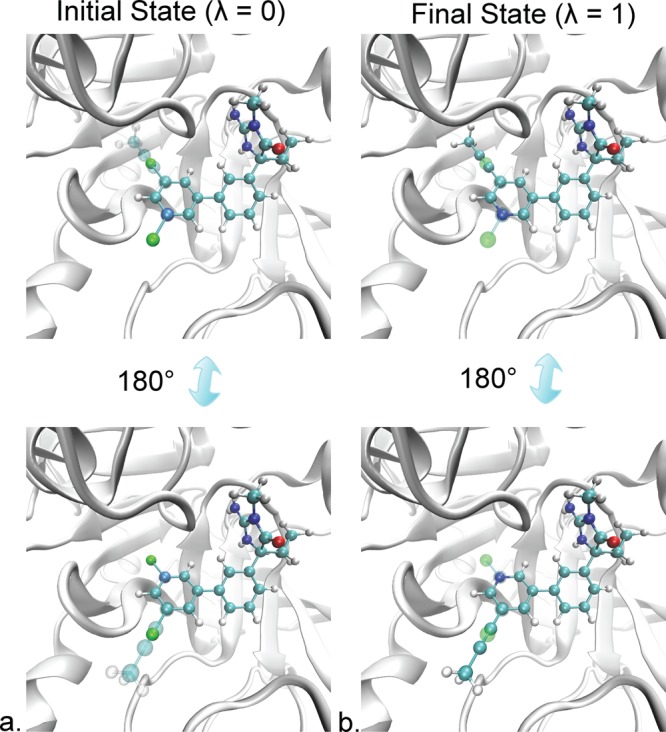
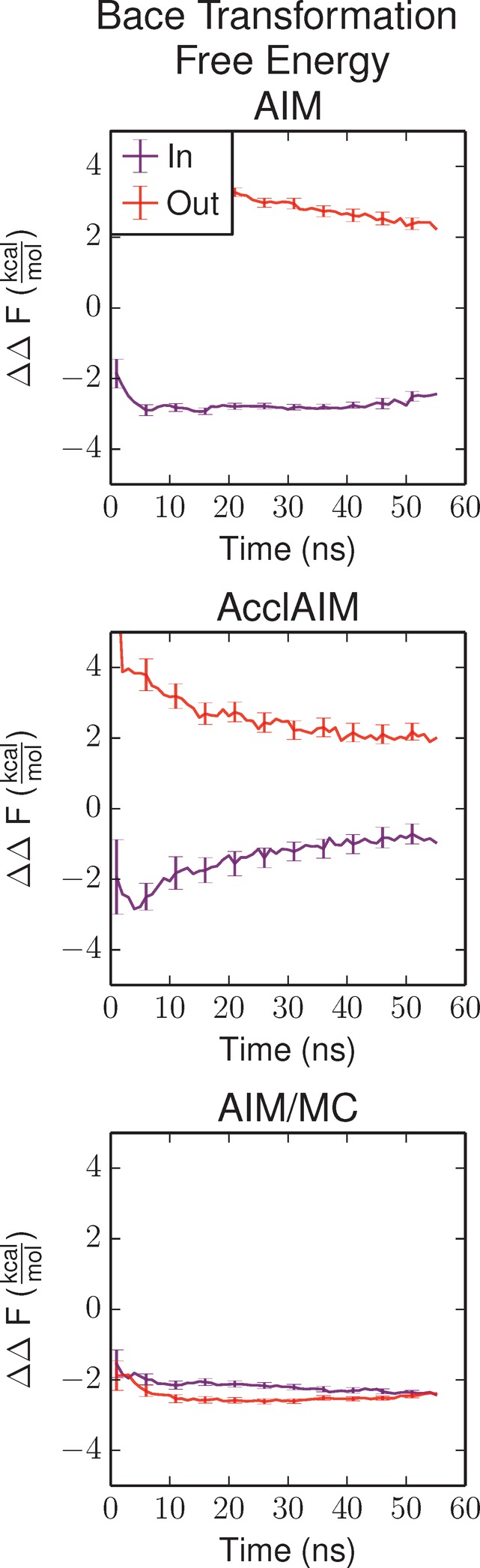
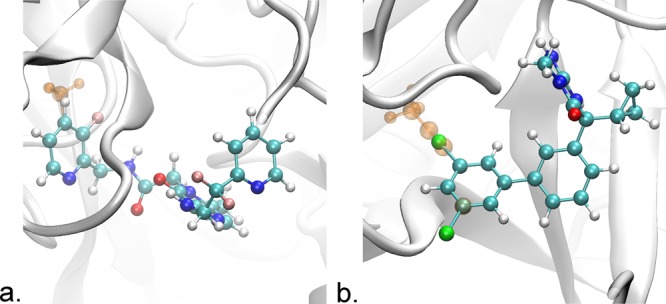
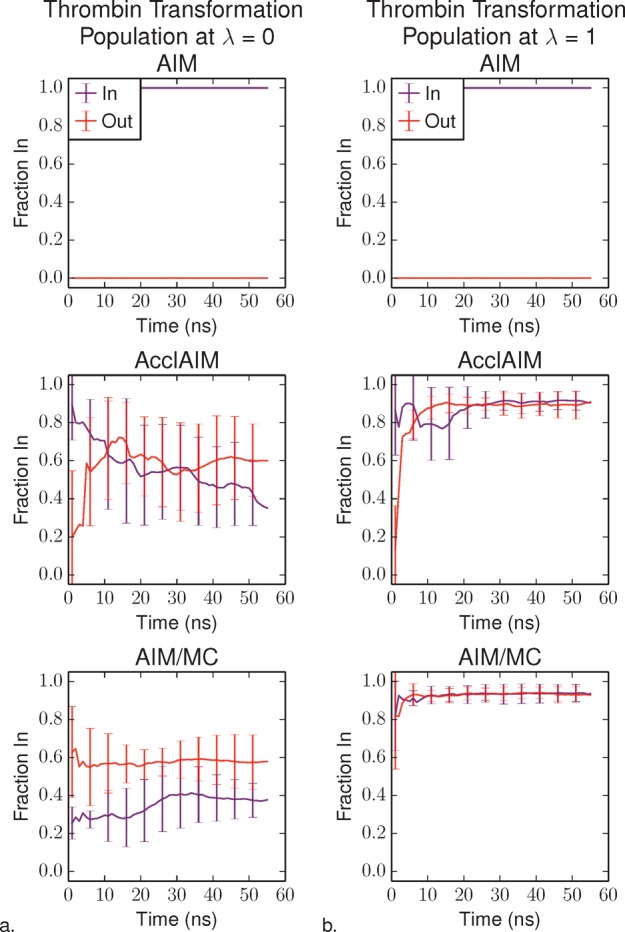
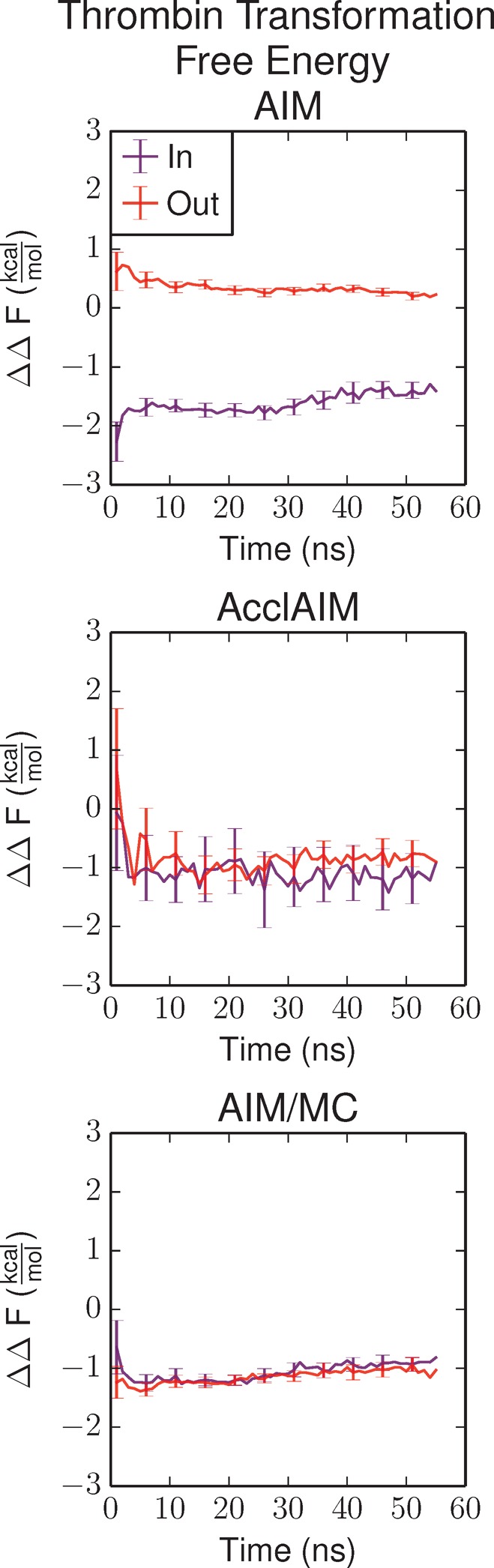
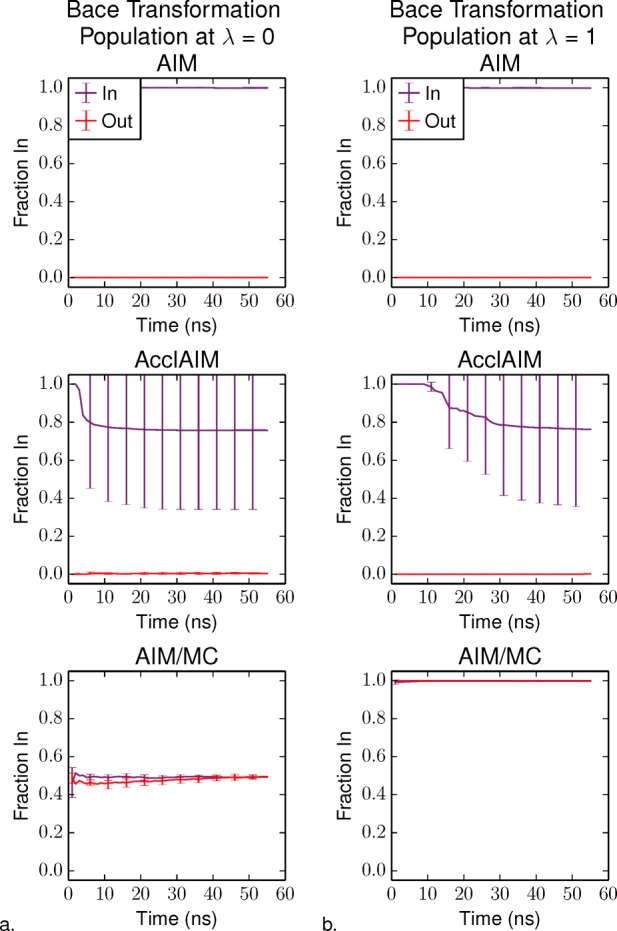
Free energy calculations are used to study how strongly potential drug molecules interact with their target receptors. The accuracy of these calculations depends on the accuracy of the molecular dynamics (MD) force field as well as proper sampling of the major conformations of each molecule. However, proper sampling of ligand conformations can be difficult when there are large barriers separating the major ligand conformations. An example of this is for ligands with an asymmetrically substituted phenyl ring, where the presence of protein loops hinders the proper sampling of the different ring conformations. These ring conformations become more difficult to sample when the size of the functional groups attached to the ring increases. The Adaptive Integration Method (AIM) has been developed, which adaptively changes the alchemical coupling parameter λ during the MD simulation so that conformations sampled at one λ can aid sampling at the other λ values. The Accelerated Adaptive Integration Method (AcclAIM) builds on AIM by lowering potential barriers for specific degrees of freedom at intermediate λ values. However, these methods may not work when there are very large barriers separating the major ligand conformations. In this work, we describe a modification to AIM that improves sampling of the different ring conformations, even when there is a very large barrier between them. This method combines AIM with conformational Monte Carlo sampling, giving improved convergence of ring populations and the resulting free energy. This method, called AIM/MC, is applied to study the relative binding free energy for a pair of ligands that bind to thrombin and a different pair of ligands that bind to aspartyl protease β-APP cleaving enzyme 1 (BACE1). These protein-ligand binding free energy calculations illustrate the improvements in conformational sampling and the convergence of the free energy compared to both AIM and AcclAIM.
自由能计算用于研究潜在药物分子与其靶受体之间的相互作用强度。这些计算的准确性取决于分子动力学(MD)力场的准确性以及每个分子主要构象的适当采样。然而,当主要配体构象之间存在较大障碍时,配体构象的适当采样可能会很困难。一个例子是具有不对称取代苯环的配体,其中蛋白质环的存在阻碍了不同环构象的适当采样。当连接到环上的官能团尺寸增加时,这些环构象变得更难采样。已经开发了自适应积分方法(AIM),它在MD模拟过程中自适应地改变炼金术耦合参数λ,以便在一个λ处采样的构象可以帮助在其他λ值处采样。加速自适应积分方法(AcclAIM)在AIM的基础上,通过降低中间λ值下特定自由度的势垒来实现。然而,当主要配体构象之间存在非常大的障碍时,这些方法可能不起作用。在这项工作中,我们描述了对AIM的一种修改,即使在不同环构象之间存在非常大的障碍时,也能改善对它们的采样。该方法将AIM与构象蒙特卡罗采样相结合,提高了环群体的收敛性以及由此产生的自由能。这种方法称为AIM/MC,用于研究一对与凝血酶结合的配体和另一对与天冬氨酸蛋白酶β-淀粉样前体蛋白裂解酶1(BACE1)结合的配体的相对结合自由能。这些蛋白质-配体结合自由能计算说明了与AIM和AcclAIM相比,在构象采样和自由能收敛方面的改进。