Liu Ruijie, Holik Aliaksei Z, Su Shian, Jansz Natasha, Chen Kelan, Leong Huei San, Blewitt Marnie E, Asselin-Labat Marie-Liesse, Smyth Gordon K, Ritchie Matthew E
Molecular Medicine Division, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia.
Stem Cells and Cancer Division, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia Department of Medical Biology, The University of Melbourne, Parkville, Victoria 3010, Australia.
Nucleic Acids Res. 2015 Sep 3;43(15):e97. doi: 10.1093/nar/gkv412. Epub 2015 Apr 29.
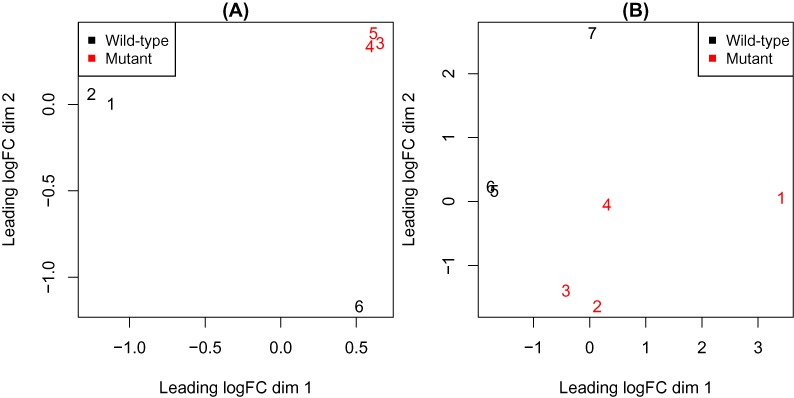
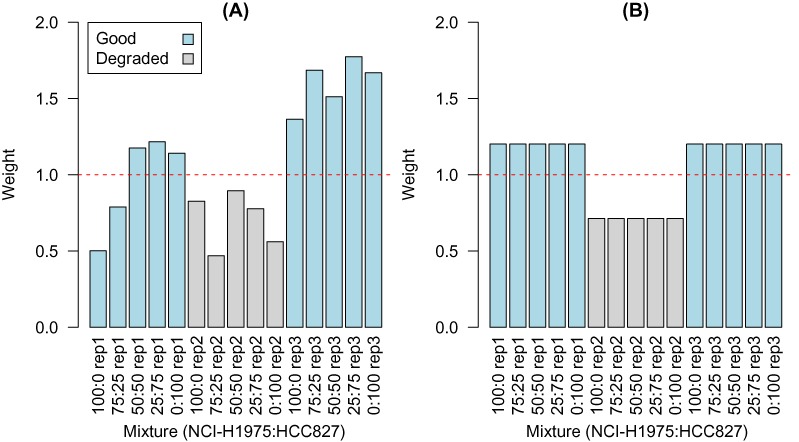
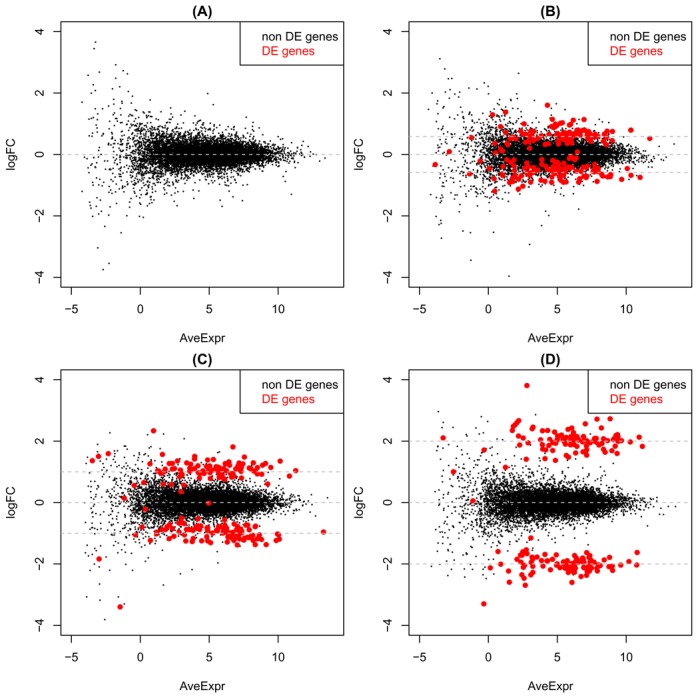
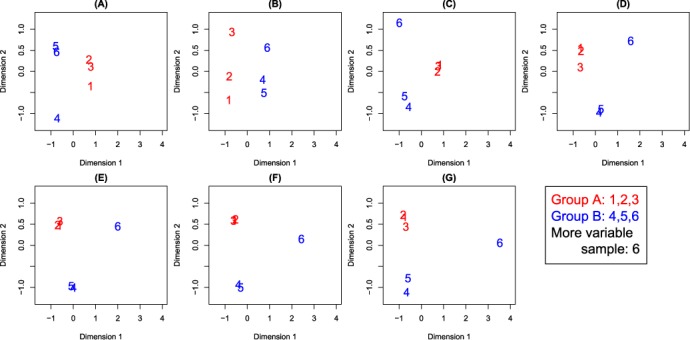
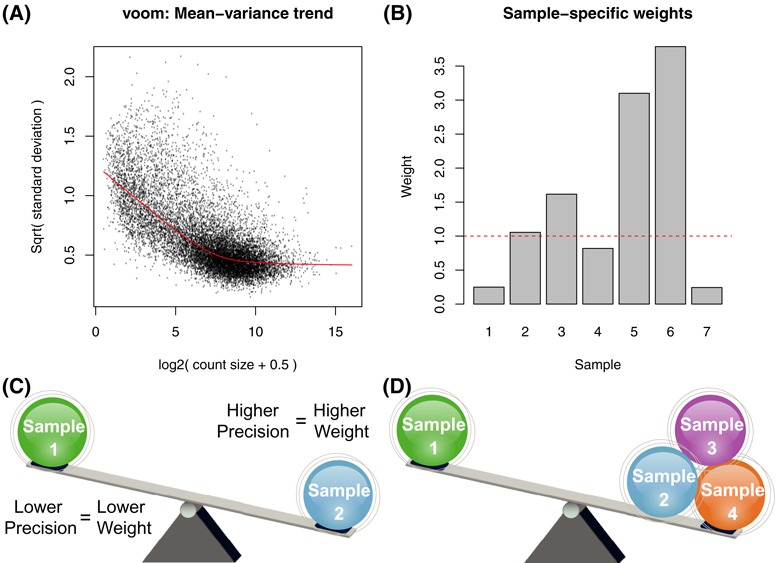
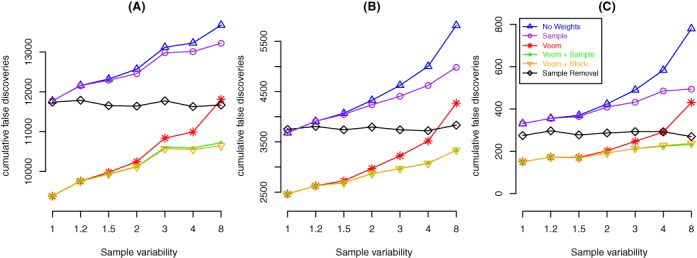
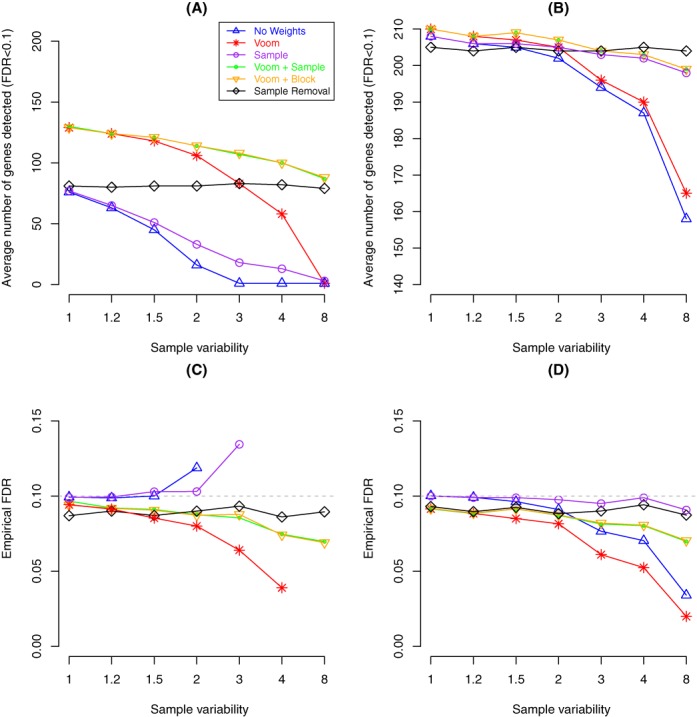
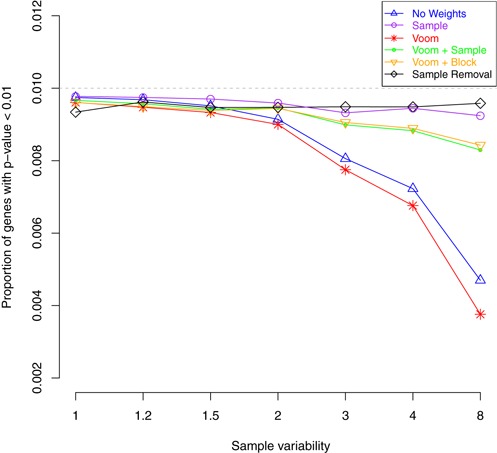
Variations in sample quality are frequently encountered in small RNA-sequencing experiments, and pose a major challenge in a differential expression analysis. Removal of high variation samples reduces noise, but at a cost of reducing power, thus limiting our ability to detect biologically meaningful changes. Similarly, retaining these samples in the analysis may not reveal any statistically significant changes due to the higher noise level. A compromise is to use all available data, but to down-weight the observations from more variable samples. We describe a statistical approach that facilitates this by modelling heterogeneity at both the sample and observational levels as part of the differential expression analysis. At the sample level this is achieved by fitting a log-linear variance model that includes common sample-specific or group-specific parameters that are shared between genes. The estimated sample variance factors are then converted to weights and combined with observational level weights obtained from the mean-variance relationship of the log-counts-per-million using 'voom'. A comprehensive analysis involving both simulations and experimental RNA-sequencing data demonstrates that this strategy leads to a universally more powerful analysis and fewer false discoveries when compared to conventional approaches. This methodology has wide application and is implemented in the open-source 'limma' package.
在小RNA测序实验中,样本质量的变化经常出现,这在差异表达分析中构成了重大挑战。去除高变异性样本可降低噪声,但代价是降低了检测能力,从而限制了我们检测生物学上有意义变化的能力。同样,在分析中保留这些样本可能由于噪声水平较高而无法揭示任何具有统计学意义的变化。一种折衷方法是使用所有可用数据,但对来自变异性更大样本的观测值进行加权。我们描述了一种统计方法,通过在差异表达分析中对样本和观测水平的异质性进行建模来实现这一点。在样本水平上,这是通过拟合一个对数线性方差模型来实现的,该模型包括基因之间共享的常见样本特异性或组特异性参数。然后将估计的样本方差因子转换为权重,并与使用“voom”从每百万对数计数的均值-方差关系获得的观测水平权重相结合。一项涉及模拟和实验RNA测序数据的综合分析表明,与传统方法相比,该策略能带来普遍更强有力的分析,且错误发现更少。这种方法具有广泛的应用,并在开源的“limma”软件包中实现。