Wang Tingting, Chen Yi-Ping Phoebe, Goddard Michael E, Meuwissen Theo H E, Kemper Kathryn E, Hayes Ben J
Faculty of Science, Technology and Engineering, La Trobe University, Melbourne, VIC, 3086, Australia.
Biosciences Research Division, Department of Primary Industries, Bundoora, Melbourne, VIC, 3083, Australia.
Genet Sel Evol. 2015 Apr 30;47(1):34. doi: 10.1186/s12711-014-0082-4.
Genomic prediction of breeding values from dense single nucleotide polymorphisms (SNP) genotypes is used for livestock and crop breeding, and can also be used to predict disease risk in humans. For some traits, the most accurate genomic predictions are achieved with non-linear estimates of SNP effects from Bayesian methods that treat SNP effects as random effects from a heavy tailed prior distribution. These Bayesian methods are usually implemented via Markov chain Monte Carlo (MCMC) schemes to sample from the posterior distribution of SNP effects, which is computationally expensive. Our aim was to develop an efficient expectation-maximisation algorithm (emBayesR) that gives similar estimates of SNP effects and accuracies of genomic prediction than the MCMC implementation of BayesR (a Bayesian method for genomic prediction), but with greatly reduced computation time.
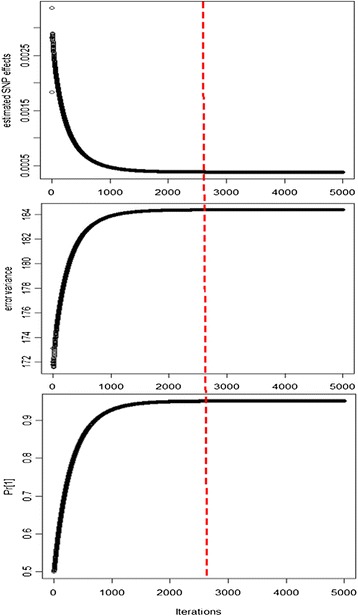
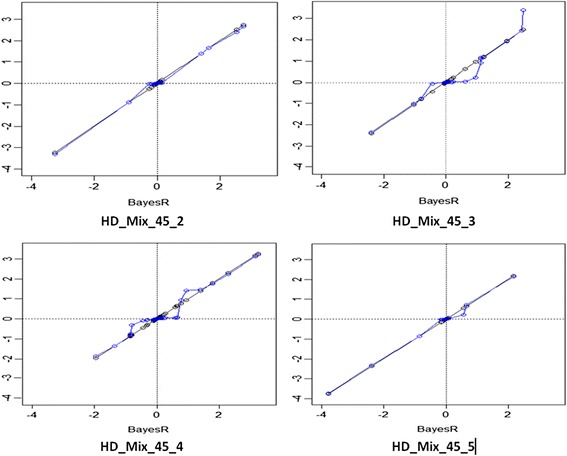
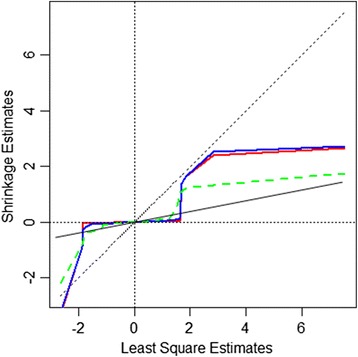
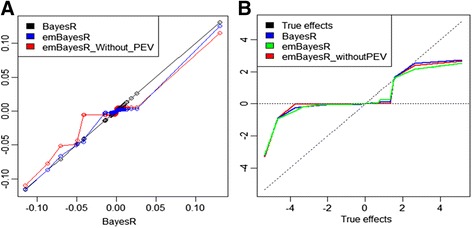
emBayesR is an approximate EM algorithm that retains the BayesR model assumption with SNP effects sampled from a mixture of normal distributions with increasing variance. emBayesR differs from other proposed non-MCMC implementations of Bayesian methods for genomic prediction in that it estimates the effect of each SNP while allowing for the error associated with estimation of all other SNP effects. emBayesR was compared to BayesR using simulated data, and real dairy cattle data with 632 003 SNPs genotyped, to determine if the MCMC and the expectation-maximisation approaches give similar accuracies of genomic prediction.
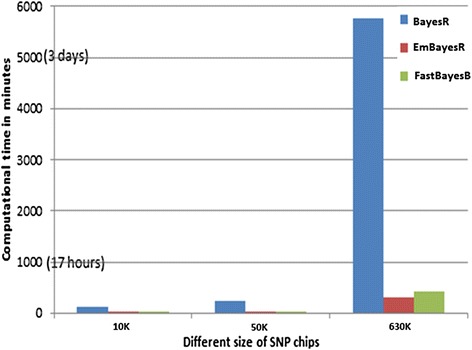
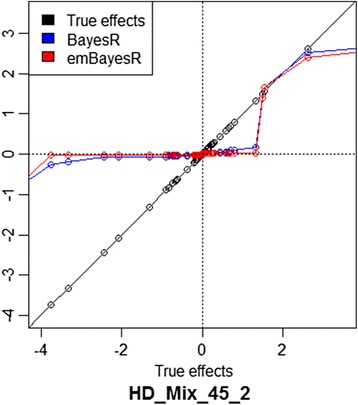
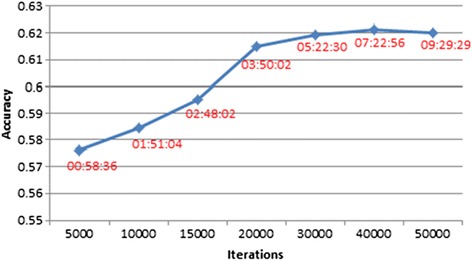
We were able to demonstrate that allowing for the error associated with estimation of other SNP effects when estimating the effect of each SNP in emBayesR improved the accuracy of genomic prediction over emBayesR without including this error correction, with both simulated and real data. When averaged over nine dairy traits, the accuracy of genomic prediction with emBayesR was only 0.5% lower than that from BayesR. However, emBayesR reduced computing time up to 8-fold compared to BayesR.
The emBayesR algorithm described here achieved similar accuracies of genomic prediction to BayesR for a range of simulated and real 630 K dairy SNP data. emBayesR needs less computing time than BayesR, which will allow it to be applied to larger datasets.
利用密集单核苷酸多态性(SNP)基因型对育种值进行基因组预测已应用于家畜和作物育种,也可用于预测人类疾病风险。对于某些性状,通过贝叶斯方法对SNP效应进行非线性估计可实现最准确的基因组预测,该方法将SNP效应视为来自重尾先验分布的随机效应。这些贝叶斯方法通常通过马尔可夫链蒙特卡罗(MCMC)方案来从SNP效应的后验分布中抽样,计算成本很高。我们的目标是开发一种高效的期望最大化算法(emBayesR),它能给出与BayesR(一种用于基因组预测的贝叶斯方法)的MCMC实现类似的SNP效应估计和基因组预测准确性,但计算时间大幅减少。
emBayesR是一种近似期望最大化算法,保留了BayesR模型假设,SNP效应从方差递增的正态分布混合中抽样。emBayesR与其他提出的用于基因组预测的贝叶斯方法的非MCMC实现的不同之处在于,它在估计每个SNP的效应时考虑了与所有其他SNP效应估计相关的误差。使用模拟数据以及对632003个SNP进行基因分型的真实奶牛数据,将emBayesR与BayesR进行比较,以确定MCMC方法和期望最大化方法是否给出相似的基因组预测准确性。
我们能够证明,在emBayesR中估计每个SNP效应时考虑与其他SNP效应估计相关的误差,相比于不包括此误差校正的emBayesR,在模拟数据和真实数据中均提高了基因组预测的准确性。对九个奶牛性状进行平均时,emBayesR的基因组预测准确性仅比BayesR低0.5%。然而,与BayesR相比,emBayesR将计算时间减少了多达8倍。
本文描述的emBayesR算法对于一系列模拟和真实的630K奶牛SNP数据,实现了与BayesR相似的基因组预测准确性。emBayesR所需的计算时间比BayesR少,这将使其能够应用于更大的数据集。