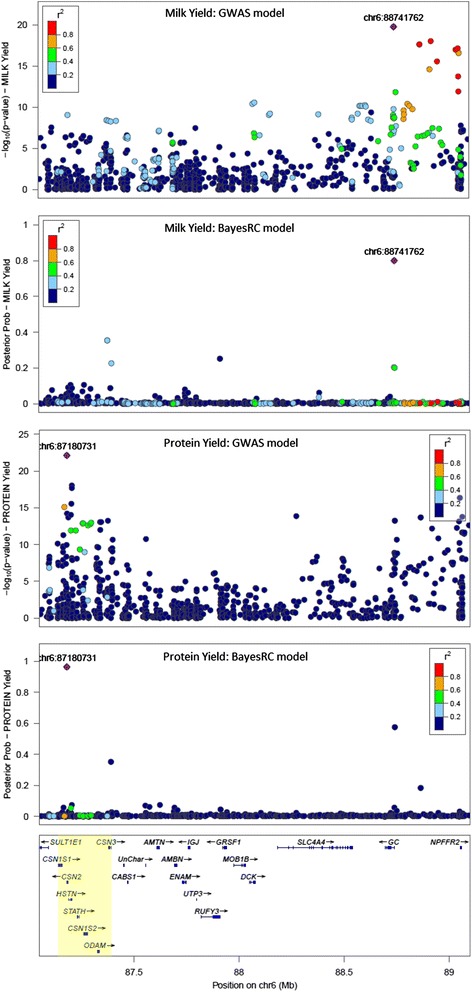
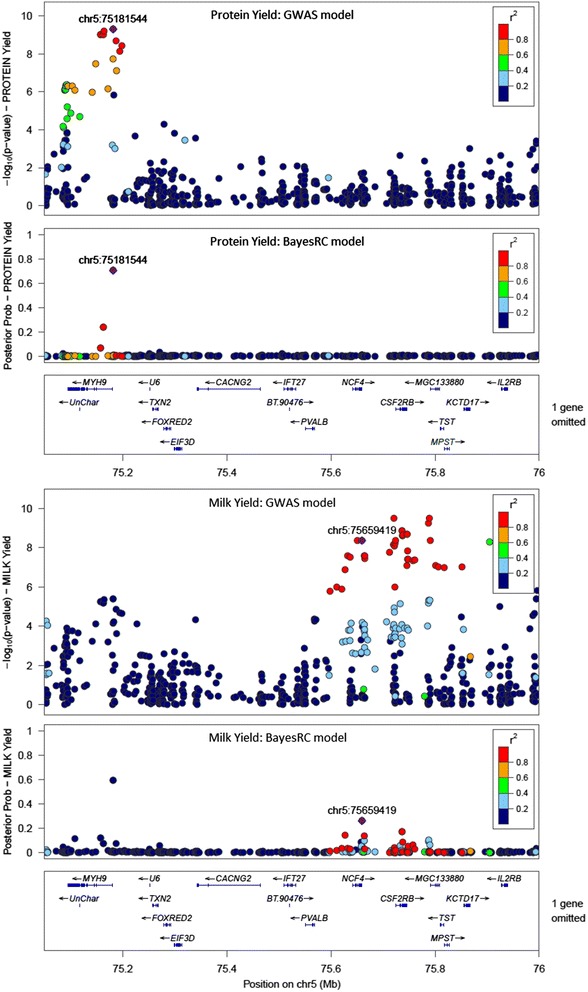
MacLeod I M, Bowman P J, Vander Jagt C J, Haile-Mariam M, Kemper K E, Chamberlain A J, Schrooten C, Hayes B J, Goddard M E
Faculty of Veterinary & Agricultural Science, University of Melbourne, Victoria, 3010, Australia.
Dairy Futures Cooperative Research Centre, AgriBio, Bundoora, Victoria, Australia.
BMC Genomics. 2016 Feb 27;17:144. doi: 10.1186/s12864-016-2443-6.
Dense SNP genotypes are often combined with complex trait phenotypes to map causal variants, study genetic architecture and provide genomic predictions for individuals with genotypes but no phenotype. A single method of analysis that jointly fits all genotypes in a Bayesian mixture model (BayesR) has been shown to competitively address all 3 purposes simultaneously. However, BayesR and other similar methods ignore prior biological knowledge and assume all genotypes are equally likely to affect the trait. While this assumption is reasonable for SNP array genotypes, it is less sensible if genotypes are whole-genome sequence variants which should include causal variants.
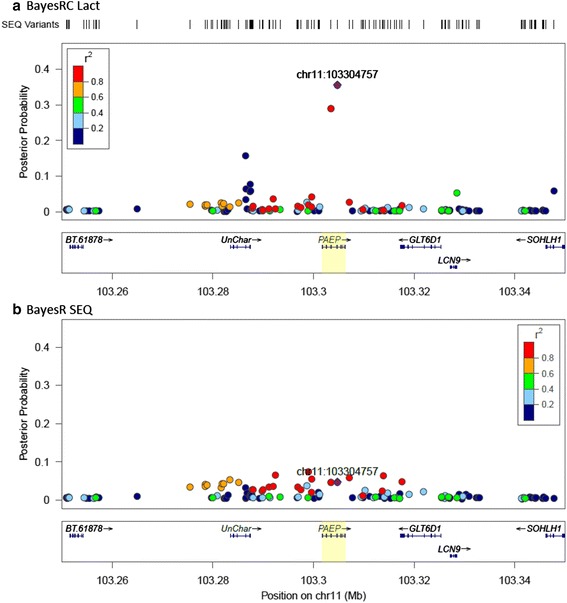
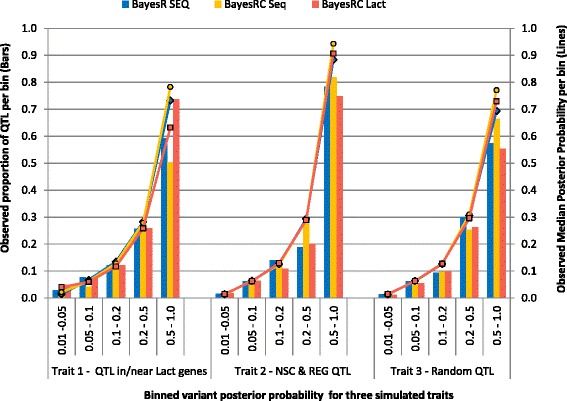
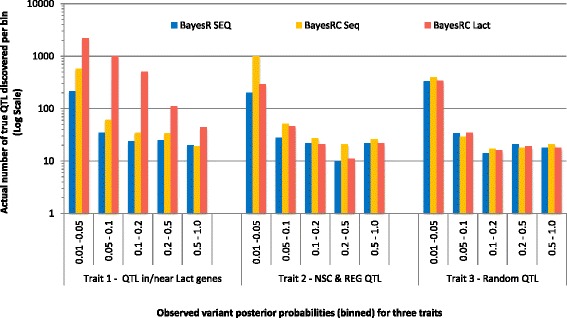
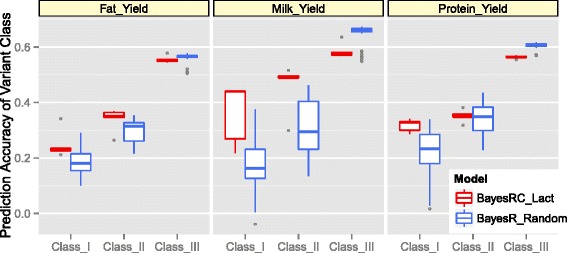
We introduce a new method (BayesRC) based on BayesR that incorporates prior biological information in the analysis by defining classes of variants likely to be enriched for causal mutations. The information can be derived from a range of sources, including variant annotation, candidate gene lists and known causal variants. This information is then incorporated objectively in the analysis based on evidence of enrichment in the data. We demonstrate the increased power of BayesRC compared to BayesR using real dairy cattle genotypes with simulated phenotypes. The genotypes were imputed whole-genome sequence variants in coding regions combined with dense SNP markers. BayesRC increased the power to detect causal variants and increased the accuracy of genomic prediction. The relative improvement for genomic prediction was most apparent in validation populations that were not closely related to the reference population. We also applied BayesRC to real milk production phenotypes in dairy cattle using independent biological priors from gene expression analyses. Although current biological knowledge of which genes and variants affect milk production is still very incomplete, our results suggest that the new BayesRC method was equal to or more powerful than BayesR for detecting candidate causal variants and for genomic prediction of milk traits.
BayesRC provides a novel and flexible approach to simultaneously improving the accuracy of QTL discovery and genomic prediction by taking advantage of prior biological knowledge. Approaches such as BayesRC will become increasing useful as biological knowledge accumulates regarding functional regions of the genome for a range of traits and species.
密集的单核苷酸多态性(SNP)基因型常与复杂性状表型相结合,以定位因果变异、研究遗传结构,并为有基因型但无表型的个体提供基因组预测。一种在贝叶斯混合模型(BayesR)中联合拟合所有基因型的单一分析方法已被证明能同时有效地实现所有这三个目标。然而,BayesR和其他类似方法忽略了先验生物学知识,并假设所有基因型影响性状的可能性相同。虽然这一假设对于SNP阵列基因型是合理的,但对于应包含因果变异的全基因组序列变异来说就不那么合理了。
我们基于BayesR引入了一种新方法(BayesRC),该方法通过定义可能富含因果突变的变异类别,在分析中纳入先验生物学信息。该信息可以从一系列来源获得,包括变异注释、候选基因列表和已知因果变异。然后,基于数据中富集的证据,将该信息客观地纳入分析。我们使用具有模拟表型的真实奶牛基因型,证明了BayesRC与BayesR相比具有更强的功效。这些基因型是编码区的推算全基因组序列变异与密集SNP标记的组合。BayesRC提高了检测因果变异的能力,并提高了基因组预测的准确性。基因组预测的相对改进在与参考群体关系不密切的验证群体中最为明显。我们还使用来自基因表达分析的独立生物学先验信息,将BayesRC应用于奶牛的实际产奶表型。尽管目前关于哪些基因和变异影响产奶的生物学知识仍然非常不完整,但我们的结果表明,新的BayesRC方法在检测候选因果变异和产奶性状的基因组预测方面与BayesR相当或更强大。
BayesRC提供了一种新颖且灵活的方法,通过利用先验生物学知识,同时提高数量性状位点(QTL)发现和基因组预测的准确性。随着关于一系列性状和物种的基因组功能区域的生物学知识不断积累,诸如BayesRC这样的方法将变得越来越有用。