Limongelli Ivan, Marini Simone, Bellazzi Riccardo
IRCCS Policlinico S. Matteo, Pzz.le Volontari del Sangue 2, 27100, Pavia, Italy.
Department of Electrical, Computer and Biomedical Engineering, University of Pavia, Via Ferrata 1, 27100, Pavia, Italy.
BMC Bioinformatics. 2015 Apr 19;16:123. doi: 10.1186/s12859-015-0554-8.
High throughput sequencing technologies are able to identify the whole genomic variation of an individual. Gene-targeted and whole-exome experiments are mainly focused on coding sequence variants related to a single or multiple nucleotides. The analysis of the biological significance of this multitude of genomic variant is challenging and computational demanding.
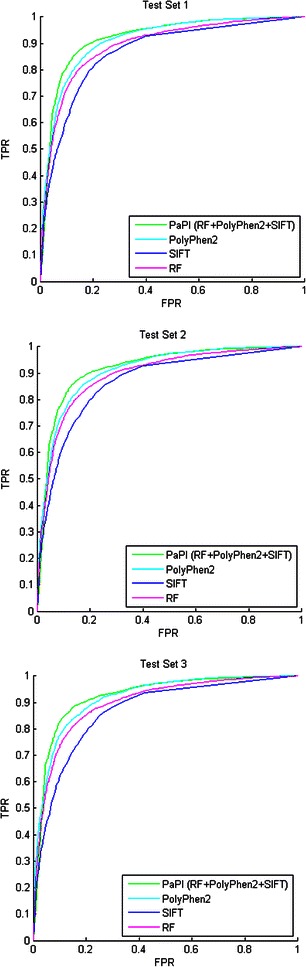
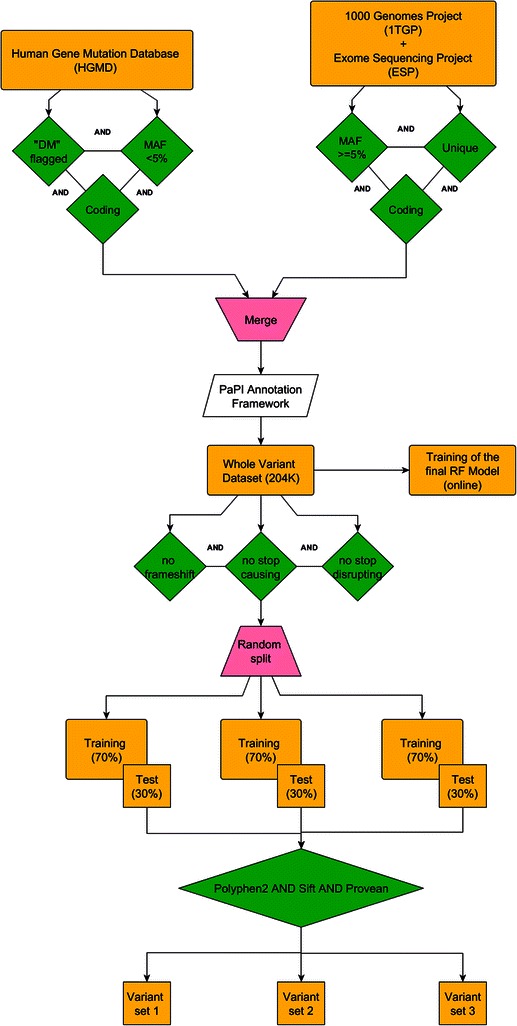
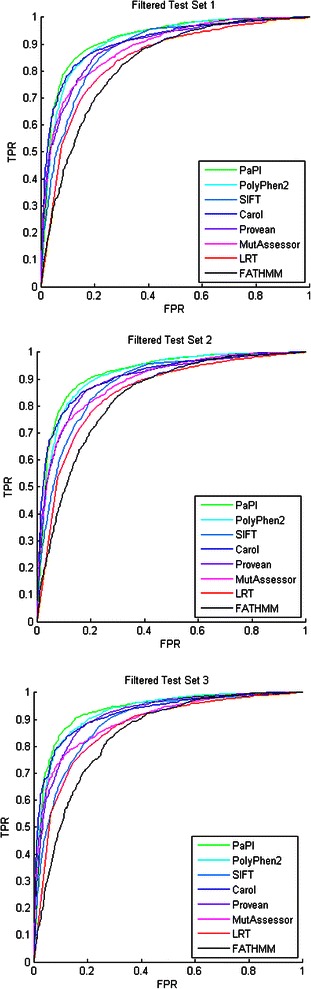
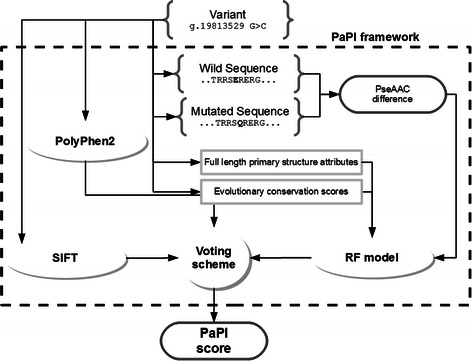
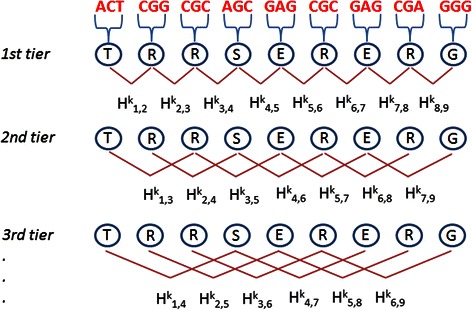
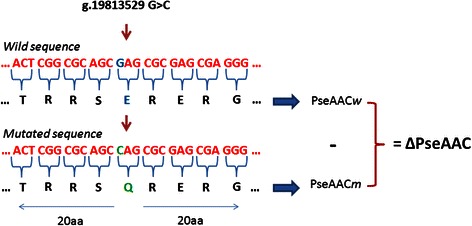
We present PaPI, a new machine-learning approach to classify and score human coding variants by estimating the probability to damage their protein-related function. The novelty of this approach consists in using pseudo amino acid composition through which wild and mutated protein sequences are represented in a discrete model. A machine learning classifier has been trained on a set of known deleterious and benign coding variants with the aim to score unobserved variants by taking into account hidden sequence patterns in human genome potentially leading to diseases. We show how the combination of amphiphilic pseudo amino acid composition, evolutionary conservation and homologous proteins based methods outperforms several prediction algorithms and it is also able to score complex variants such as deletions, insertions and indels.
This paper describes a machine-learning approach to predict the deleteriousness of human coding variants. A freely available web application (http://papi.unipv.it) has been developed with the presented method, able to score up to thousands variants in a single run.
高通量测序技术能够识别个体的全基因组变异。基因靶向和全外显子实验主要聚焦于与单个或多个核苷酸相关的编码序列变异。分析如此众多基因组变异的生物学意义具有挑战性且对计算要求很高。
我们提出了PaPI,一种通过估计损害其蛋白质相关功能的概率来对人类编码变异进行分类和评分的新机器学习方法。该方法的新颖之处在于使用伪氨基酸组成,通过它野生型和突变型蛋白质序列以离散模型表示。一个机器学习分类器已在一组已知的有害和良性编码变异上进行训练,目的是通过考虑人类基因组中可能导致疾病的隐藏序列模式来对未观察到的变异进行评分。我们展示了两亲性伪氨基酸组成、进化保守性和基于同源蛋白质的方法相结合如何优于几种预测算法,并且它还能够对缺失、插入和插入缺失等复杂变异进行评分。
本文描述了一种预测人类编码变异有害性的机器学习方法。已使用所提出的方法开发了一个免费的网络应用程序(http://papi.unipv.it),能够在单次运行中对多达数千个变异进行评分。