Interuniversity Institute of Bioinformatics in Brussels, ULB/VUB, Triomflaan, BC building, 6th floor, CP 263, 1050 Brussels, Belgium.
Machine Learning Group, Université Libre de Bruxelles, Boulevard du Triomphe, CP 212, 1050 Brussels, Belgium.
Nucleic Acids Res. 2017 Jul 3;45(W1):W201-W206. doi: 10.1093/nar/gkx390.
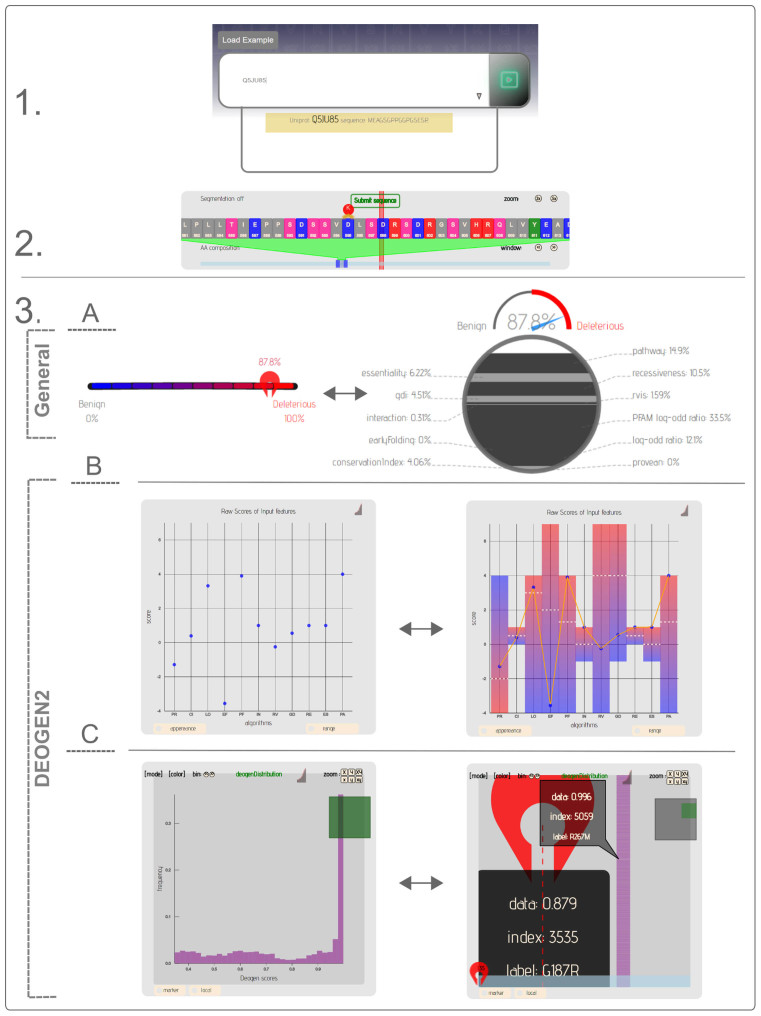
High-throughput sequencing methods are generating enormous amounts of genomic data, giving unprecedented insights into human genetic variation and its relation to disease. An individual human genome contains millions of Single Nucleotide Variants: to discriminate the deleterious from the benign ones, a variety of methods have been developed that predict whether a protein-coding variant likely affects the carrier individual's health. We present such a method, DEOGEN2, which incorporates heterogeneous information about the molecular effects of the variants, the domains involved, the relevance of the gene and the interactions in which it participates. This extensive contextual information is non-linearly mapped into one single deleteriousness score for each variant. Since for the non-expert user it is sometimes still difficult to assess what this score means, how it relates to the encoded protein, and where it originates from, we developed an interactive online framework (http://deogen2.mutaframe.com/) to better present the DEOGEN2 deleteriousness predictions of all possible variants in all human proteins. The prediction is visualized so both expert and non-expert users can gain insights into the meaning, protein context and origins of each prediction.
高通量测序方法正在产生大量的基因组数据,使人们对人类遗传变异及其与疾病的关系有了前所未有的了解。一个人类基因组包含数百万个单核苷酸变异:为了区分有害和良性的变异,已经开发了多种方法来预测编码蛋白变异是否可能影响携带者的健康。我们提出了这样一种方法,即 DEOGEN2,它结合了关于变异的分子效应、涉及的结构域、基因相关性以及它参与的相互作用的异构信息。这种广泛的上下文信息被非线性地映射到每个变异的单一有害性评分中。由于对于非专业用户来说,有时仍然很难评估这个评分意味着什么,它与编码蛋白的关系,以及它的来源,我们开发了一个交互式在线框架(http://deogen2.mutaframe.com/),以便更好地展示 DEOGEN2 在所有人类蛋白中所有可能变异的有害性预测。该预测以可视化的方式呈现,以便专家和非专业用户都可以深入了解每个预测的含义、蛋白背景和来源。