Nagy Peter I
Center for Drug Design and Development, the University of Toledo, Toledo, OH 43606, USA.
Int J Mol Sci. 2015 May 13;16(5):10767-96. doi: 10.3390/ijms160510767.
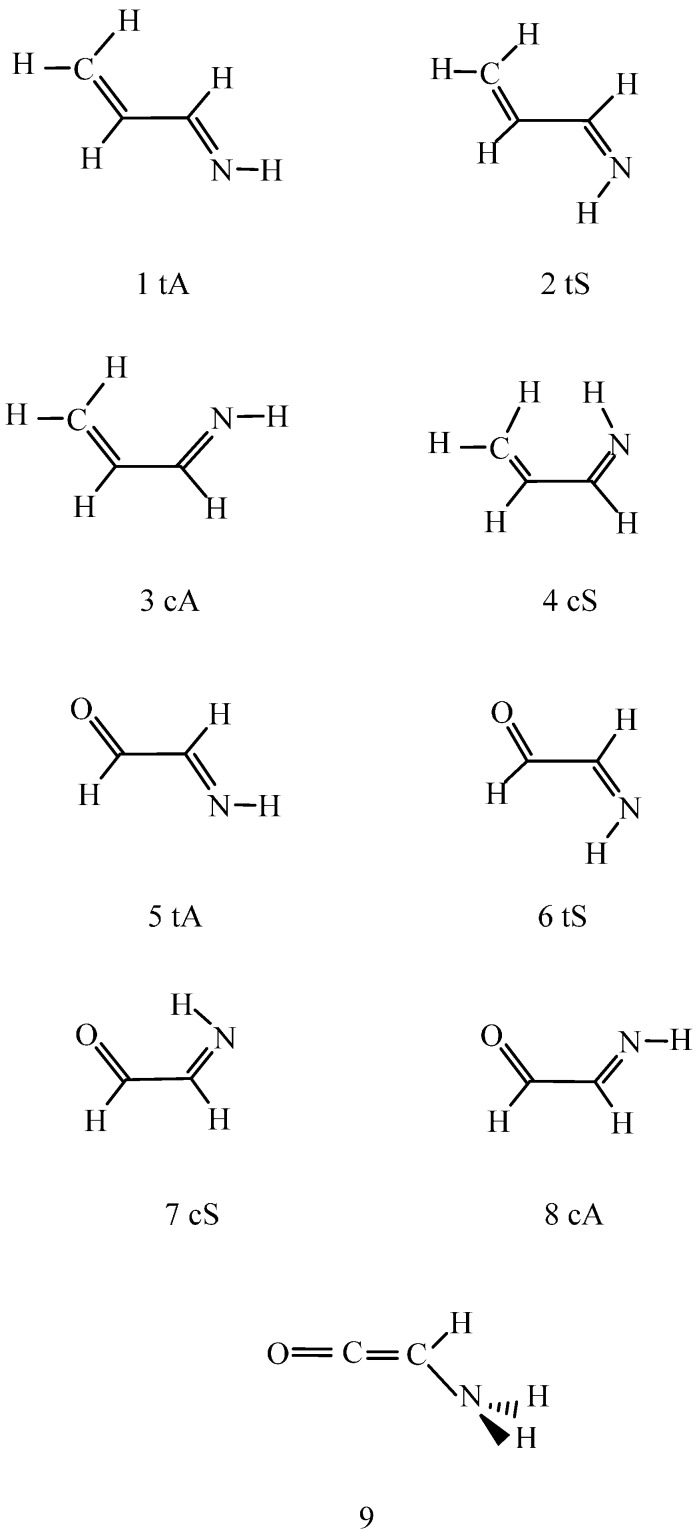
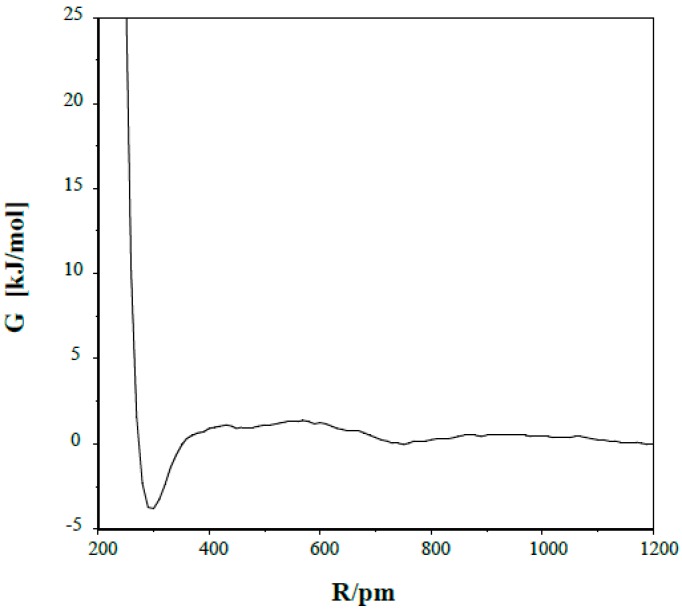
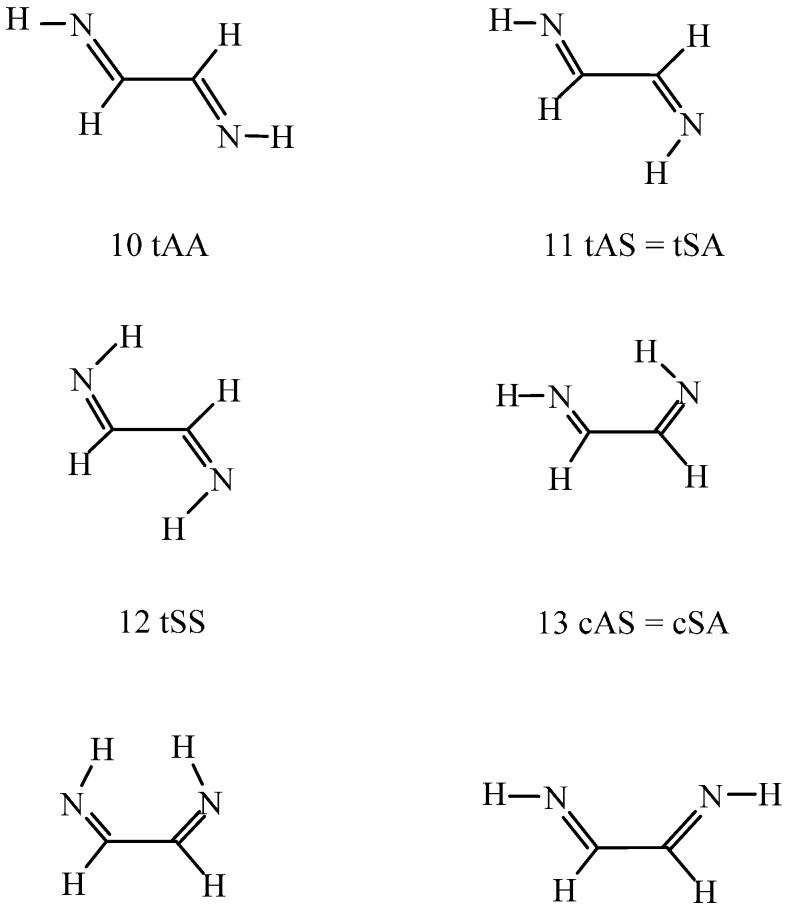
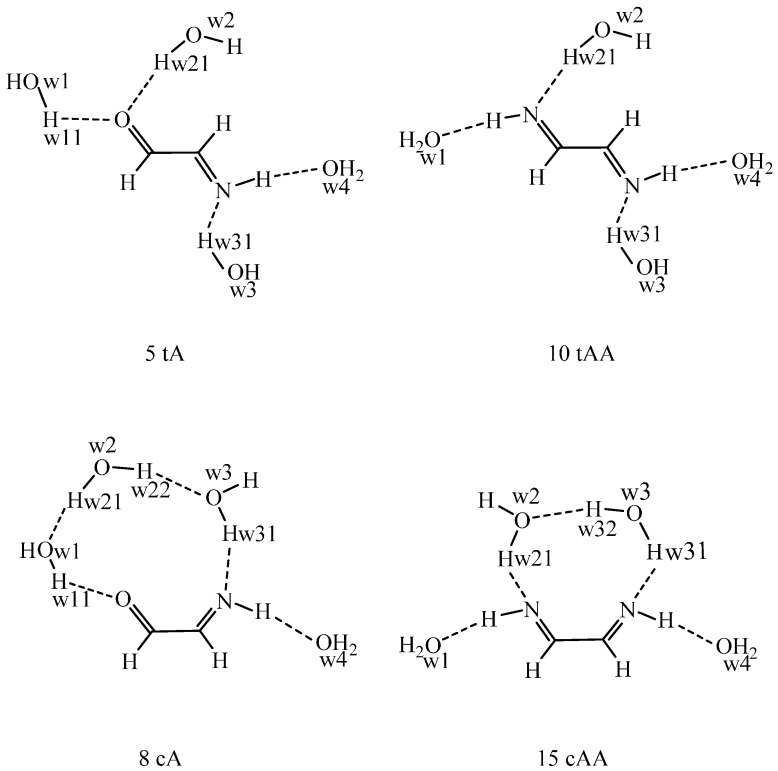
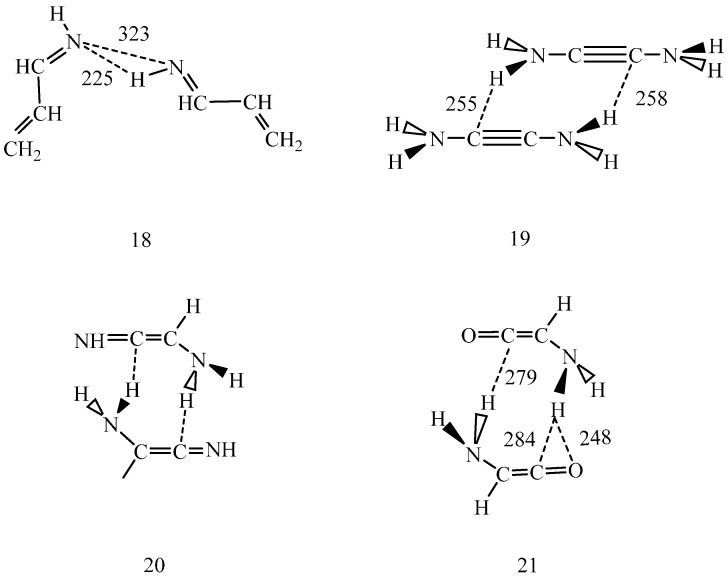
Conformational/tautomeric transformations for X=CH-CH=Y structures (X = CH2, O, NH and Y = NH) have been studied in the gas phase, in dichloromethane and in aqueous solutions. The paper is a continuation of a former study where s-cis/s-trans conformational equilibria were predicted for analogues. The s-trans conformation is preferred for the present molecules in the gas phase on the basis of its lowest internal free energy as calculated at the B97D/aug-cc-pvqz and CCSD(T)CBS (coupled-cluster singles and doubles with non-iterative triples extrapolated to the complete basis set) levels. Transition state barriers are of 29-36 kJ/mol for rotations about the central C-C bonds. In solution, an s-trans form is still favored on the basis of its considerably lower internal free energy compared with the s-cis forms as calculated by IEF-PCM (integral-equation formalism of the polarizable continuum dielectric solvent model) at the theoretical levels indicated. A tetrahydrate model in the supermolecule/continuum approach helped explore the 2solute-solvent hydrogen bond pattern. The calculated transition state barrier for rotation about the C-C bond decreased to 27 kJ/mol for the tetrahydrate. Considering explicit solvent models, relative solvation free energies were calculated by means of the free energy perturbation method through Monte Carlo simulations. These calculated values differ remarkably from those by the PCM approach in aqueous solution, nonetheless the same prevalent conformation was predicted by the two methods. Aqueous solution structure-characteristics were determined by Monte Carlo. Equilibration of conformers/tautomers through water-assisted double proton-relay is discussed. This mechanism is not viable, however, in non-protic solvents where the calculated potential of mean force curve does not predict remarkable solute dimerization and subsequent favorable orientation.
已在气相、二氯甲烷和水溶液中研究了X=CH-CH=Y结构(X = CH₂、O、NH且Y = NH)的构象/互变异构转变。本文是前一项研究的延续,在前一项研究中预测了类似物的s-顺式/s-反式构象平衡。基于在B97D/aug-cc-pvqz和CCSD(T)CBS(耦合簇单双激发并外推非迭代三激发至完整基组)水平计算出的最低内部自由能,在气相中当前分子更倾向于s-反式构象。围绕中心C-C键旋转的过渡态能垒为29 - 36 kJ/mol。在溶液中,基于通过IEF-PCM(可极化连续介质介电溶剂模型的积分方程形式)在所示理论水平计算得出的s-反式形式与s-顺式形式相比显著更低的内部自由能,s-反式形式仍然更受青睐。超分子/连续介质方法中的四水合物模型有助于探索2溶质 - 溶剂氢键模式。对于四水合物,围绕C-C键旋转的计算过渡态能垒降至27 kJ/mol。考虑显式溶剂模型,通过蒙特卡罗模拟利用自由能微扰法计算了相对溶剂化自由能。这些计算值与水溶液中PCM方法得到的值显著不同,尽管两种方法预测的优势构象相同。通过蒙特卡罗确定了水溶液的结构特征。讨论了通过水辅助双质子传递实现构象异构体/互变异构体的平衡。然而,在非质子溶剂中这种机制不可行,在非质子溶剂中计算出的平均力势曲线未预测到显著的溶质二聚化及随后的有利取向。