Takumi Katsuhisa, Cacciò Simone M, van der Giessen Joke, Xiao Lihua, Sprong Hein
Centre for Infectious Disease Control, National Institute for Public Health and the Environment (RIVM), P.O. Box 1, Bilthoven, 3720, The Netherlands.
Department of Infectious, Parasitic and Immunomediated Diseases, Istituto Superiore di Sanità, Rome, Italy.
Parasit Vectors. 2015 Jun 6;8:308. doi: 10.1186/s13071-015-0921-3.
Cryptosporidiosis is a gastrointestinal disease affecting many people worldwide. Disease incidence is often unknown and surveillance of human cryptosporidiosis is installed in only a handful of developed countries. A genetic marker that mirrors disease incidence is potentially a powerful tool for monitoring the two primary human infected species of Cryptosporidium.
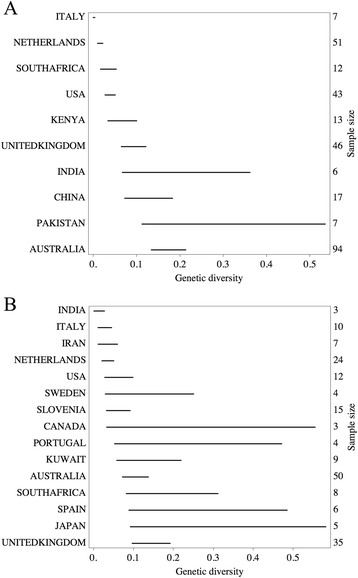
We used the molecular epidemiological database with Cryptosporidium isolates from ZoopNet, which currently contains more than 1400 records with their sampling nations, and the names of the host species from which the isolates were obtained. Based on 296 C. hominis and 195 C. parvum GP60 sequences from human origin, the genetic diversities of Cryptosporidium was estimated for several nations. Notified cases of human cryptosporidiosis were collected from statistics databases for only four nations.
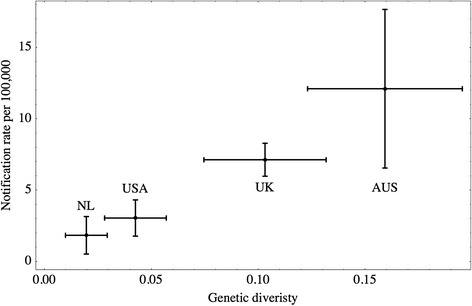
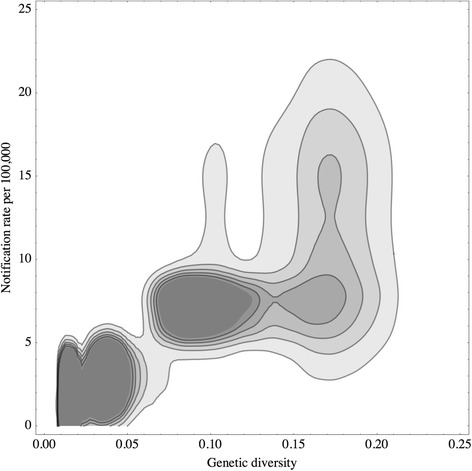
Genetic diversities of C. hominis were estimated in 10 nations in 5 continents, and that of C. parvum of human origin were estimated in 15 nations. Correlation with reported incidence of human cryptosporidiosis in four nations (the Netherlands, United States, United Kingdom and Australia) was positive and significant. A linear model for testing the relationship between the genetic diversity and incidence produced a significantly positive estimate for the slope (P-value < 0.05).
The hypothesis that genetic diversity at GP60 locus mirrors notification rates of human cryptosporidiosis was not rejected based on the data presented. Genetic diversity of C. hominis and C. parvum may therefore be an independent and complementary measure for quantifying disease incidence, for which only a moderate number of stool samples from each nation are sufficient data input.
隐孢子虫病是一种影响全球许多人的胃肠道疾病。疾病发病率通常未知,只有少数发达国家开展了人类隐孢子虫病监测。一种反映疾病发病率的基因标记可能是监测两种主要人类感染的隐孢子虫物种的有力工具。
我们使用了来自ZoopNet的隐孢子虫分离株分子流行病学数据库,该数据库目前包含1400多条记录,记录了采样国家以及分离株所获宿主物种的名称。基于296条来自人类的人隐孢子虫和195条微小隐孢子虫GP60序列,估计了几个国家隐孢子虫的遗传多样性。仅从四个国家的统计数据库收集了人类隐孢子虫病的通报病例。
估计了五大洲10个国家的人隐孢子虫遗传多样性,以及15个国家的人源微小隐孢子虫遗传多样性。与四个国家(荷兰、美国、英国和澳大利亚)报告的人类隐孢子虫病发病率呈正相关且具有显著性。用于检验遗传多样性与发病率之间关系的线性模型得出斜率的估计值显著为正(P值<0.05)。
基于所提供的数据,未拒绝GP60位点的遗传多样性反映人类隐孢子虫病通报率这一假设。因此,人隐孢子虫和微小隐孢子虫的遗传多样性可能是量化疾病发病率的一种独立且互补的指标,每个国家仅需适量数量的粪便样本作为数据输入即可。