Smedley Damian, Robinson Peter N
Skarnes Faculty Group, Wellcome Trust Sanger Institute, Hinxton, UK.
Institute for Medical Genetics and Human Genetics, Charité-Universitätsmedizin Berlin, Berlin, Germany ; Max Planck Institute for Molecular Genetics, Ihnestrasse, 14195 Berlin, Germany ; Berlin Brandenburg Center for Regenerative Therapies (BCRT), Charité-Universitätsmedizin Berlin, Augustenburger Platz, 13353 Berlin, Germany ; Institute for Bioinformatics, Department of Mathematics and Computer Science, Freie Universität Berlin, Takustrasse, 14195 Berlin, Germany.
Genome Med. 2015 Jul 30;7(1):81. doi: 10.1186/s13073-015-0199-2. eCollection 2015.
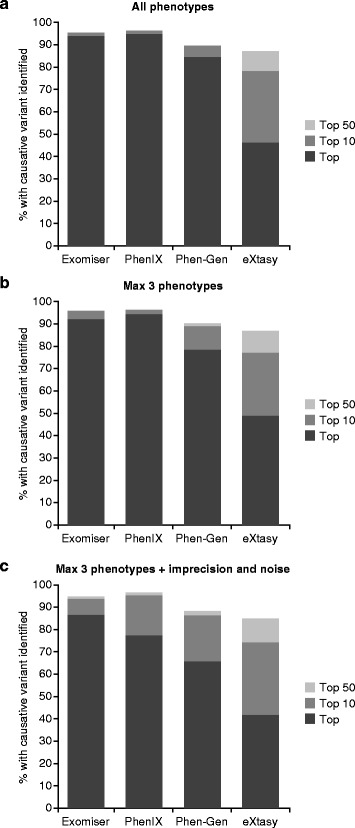
Whole exome sequencing has altered the way in which rare diseases are diagnosed and disease genes identified. Hundreds of novel disease-associated genes have been characterized by whole exome sequencing in the past five years, yet the identification of disease-causing mutations is often challenging because of the large number of rare variants that are being revealed. Gene prioritization aims to rank the most probable candidate genes towards the top of a list of potentially pathogenic variants. A promising new approach involves the computational comparison of the phenotypic abnormalities of the individual being investigated with those previously associated with human diseases or genetically modified model organisms. In this review, we compare and contrast the strengths and weaknesses of current phenotype-driven computational algorithms, including Phevor, Phen-Gen, eXtasy and two algorithms developed by our groups called PhenIX and Exomiser. Computational phenotype analysis can substantially improve the performance of exome analysis pipelines.
全外显子组测序改变了罕见病的诊断方式以及疾病基因的识别方式。在过去五年中,通过全外显子组测序已经鉴定出数百个新的疾病相关基因,然而由于大量罕见变异的出现,致病突变的识别往往具有挑战性。基因优先级排序旨在将最有可能的候选基因排在潜在致病变异列表的首位。一种有前景的新方法涉及将被研究个体的表型异常与先前与人类疾病或基因改造模式生物相关的表型异常进行计算比较。在本综述中,我们比较并对比了当前基于表型的计算算法的优缺点,包括Phevor、Phen-Gen、eXtasy以及我们团队开发的两种算法PhenIX和Exomiser。计算表型分析可以显著提高外显子组分析流程的性能。