Mutlak Michael, Kehat Izhak
The Rappaport Institute and the Bruce Rappaport Faculty of Medicine, Technion - Israel Institute of Technology , Haifa, Israel.
The Rappaport Institute and the Bruce Rappaport Faculty of Medicine, Technion - Israel Institute of Technology , Haifa, Israel ; Department of Cardiology and the Clinical Research Institute at Rambam, Rambam Medical Center , Haifa, Israel.
Front Pharmacol. 2015 Jul 24;6:149. doi: 10.3389/fphar.2015.00149. eCollection 2015.
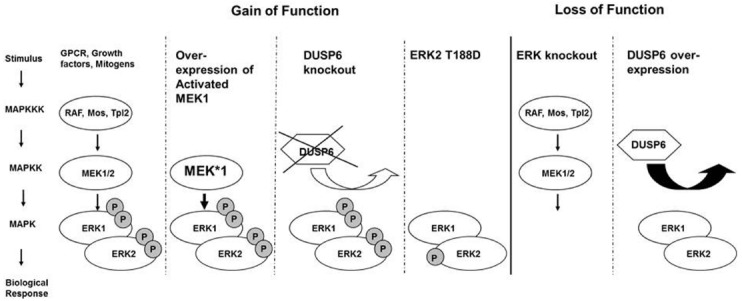
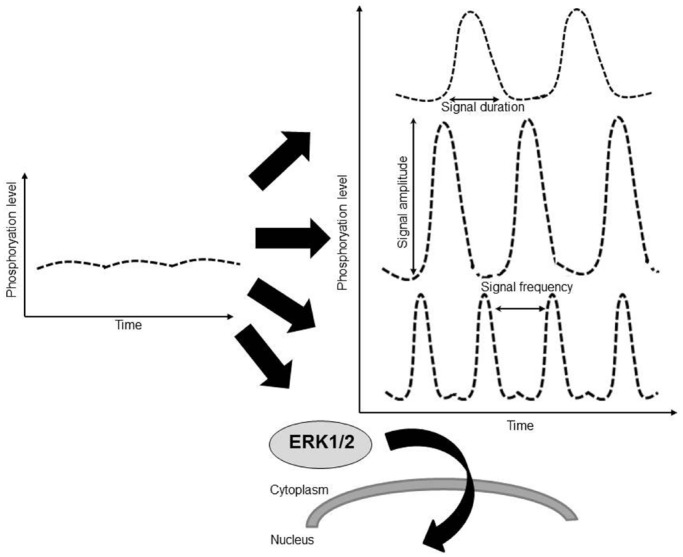
Cardiac hypertrophy results from increased mechanical load on the heart and through the actions of local and systemic neuro-humoral factors, cytokines and growth factors. These mechanical and neuroendocrine effectors act through stretch, G protein-coupled receptors and tyrosine kinases to induce the activation of a myriad of intracellular signaling pathways including the extracellular signal-regulated kinases 1/2 (ERK1/2). Since most stimuli that provoke myocardial hypertrophy also elicit an acute phosphorylation of the threonine-glutamate-tyrosine (TEY) motif within the activation loops of ERK1 and ERK2 kinases, resulting in their activation, ERKs have long been considered promotors of cardiac hypertrophy. Several mouse models were generated in order to directly understand the causal role of ERK1/2 activation in the heart. These models include direct manipulation of ERK1/2 such as overexpression, mutagenesis or knockout models, manipulations of upstream kinases such as MEK1 and manipulations of the phosphatases that dephosphorylate ERK1/2 such as DUSP6. The emerging understanding from these studies, as will be discussed here, is more complex than originally considered. While there is little doubt that ERK1/2 activation or the lack of it modulates the hypertrophic process or the type of hypertrophy that develops, it appears that not all ERK1/2 activation events are the same. While much has been learned, some questions remain regarding the exact role of ERK1/2 in the heart, the upstream events that result in ERK1/2 activation and the downstream effector in hypertrophy.
心肌肥大是由心脏机械负荷增加以及局部和全身神经体液因子、细胞因子和生长因子的作用引起的。这些机械和神经内分泌效应器通过拉伸、G蛋白偶联受体和酪氨酸激酶发挥作用,以诱导包括细胞外信号调节激酶1/2(ERK1/2)在内的众多细胞内信号通路的激活。由于大多数引发心肌肥大的刺激也会引起ERK1和ERK2激酶激活环内苏氨酸-谷氨酸-酪氨酸(TEY)基序的急性磷酸化,从而导致它们的激活,ERK长期以来一直被认为是心肌肥大的促进因子。为了直接了解ERK1/2激活在心脏中的因果作用,人们构建了几种小鼠模型。这些模型包括对ERK1/2的直接操作,如过表达、诱变或敲除模型,对上游激酶如MEK1的操作,以及对使ERK1/2去磷酸化的磷酸酶如DUSP6的操作。正如本文将要讨论的,这些研究中逐渐形成的认识比最初认为的更为复杂。虽然毫无疑问ERK1/2的激活或缺乏会调节肥大过程或所发生的肥大类型,但似乎并非所有的ERK1/2激活事件都是相同的。虽然已经了解了很多,但关于ERK1/2在心脏中的确切作用、导致ERK1/2激活的上游事件以及肥大中的下游效应器,仍有一些问题存在。