Rosenberger Albert, Friedrichs Stefanie, Amos Christopher I, Brennan Paul, Fehringer Gordon, Heinrich Joachim, Hung Rayjean J, Muley Thomas, Müller-Nurasyid Martina, Risch Angela, Bickeböller Heike
Department of Genetic Epidemiology, University Medical Center, Georg-August University Göttingen, Göttingen, Germany.
Geisel School of Medicine, Dartmouth College, Lebanon, NH, United States of America.
PLoS One. 2015 Oct 26;10(10):e0140179. doi: 10.1371/journal.pone.0140179. eCollection 2015.
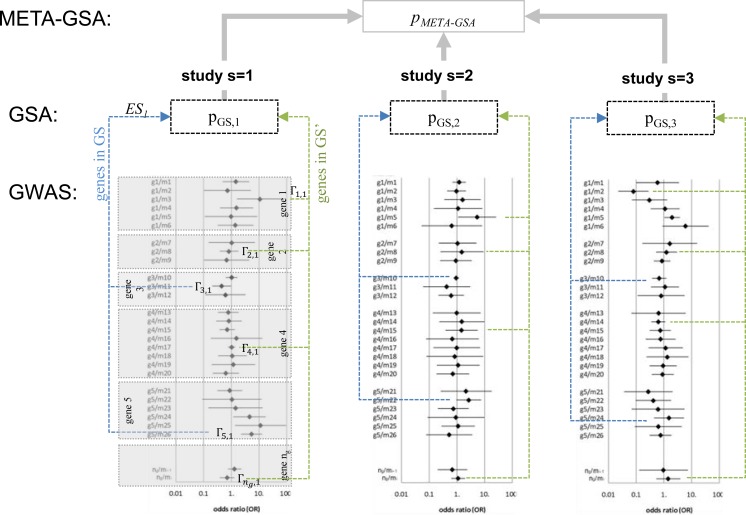
Gene-set analysis (GSA) methods are used as complementary approaches to genome-wide association studies (GWASs). The single marker association estimates of a predefined set of genes are either contrasted with those of all remaining genes or with a null non-associated background. To pool the p-values from several GSAs, it is important to take into account the concordance of the observed patterns resulting from single marker association point estimates across any given gene set. Here we propose an enhanced version of Fisher's inverse χ2-method META-GSA, however weighting each study to account for imperfect correlation between association patterns.
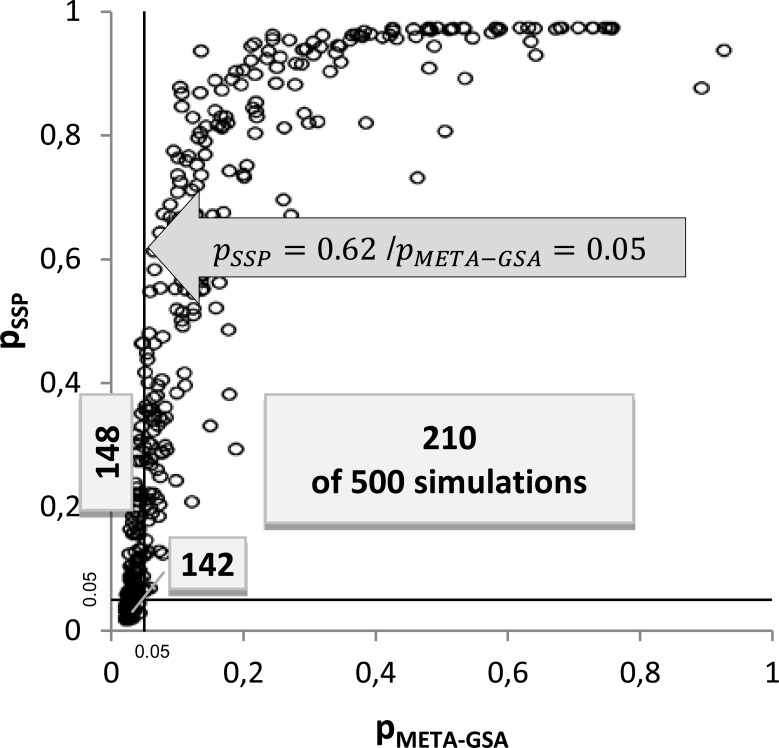
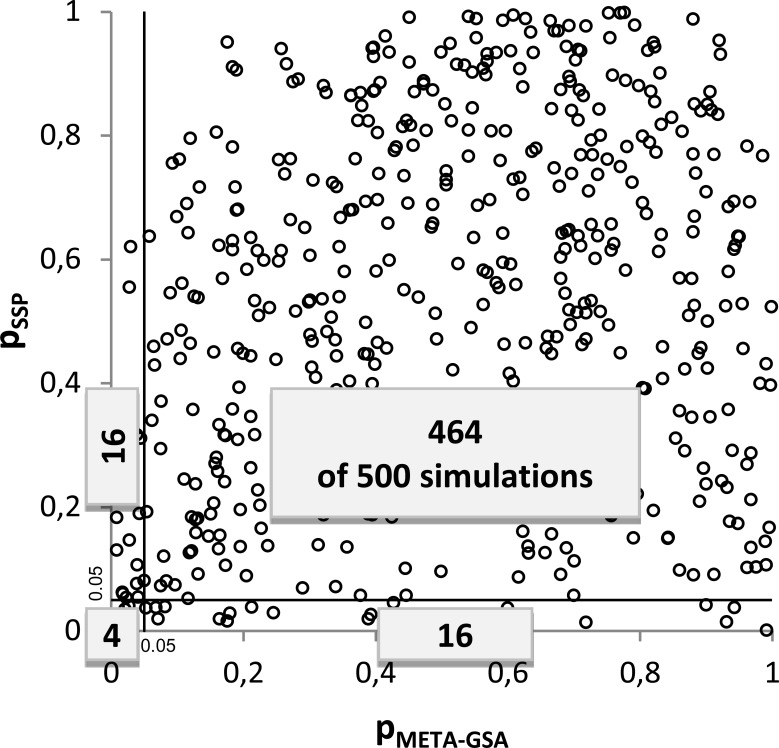
We investigated the performance of META-GSA by simulating GWASs with 500 cases and 500 controls at 100 diallelic markers in 20 different scenarios, simulating different relative risks between 1 and 1.5 in gene sets of 10 genes. Wilcoxon's rank sum test was applied as GSA for each study. We found that META-GSA has greater power to discover truly associated gene sets than simple pooling of the p-values, by e.g. 59% versus 37%, when the true relative risk for 5 of 10 genes was assume to be 1.5. Under the null hypothesis of no difference in the true association pattern between the gene set of interest and the set of remaining genes, the results of both approaches are almost uncorrelated. We recommend not relying on p-values alone when combining the results of independent GSAs.
We applied META-GSA to pool the results of four case-control GWASs of lung cancer risk (Central European Study and Toronto/Lunenfeld-Tanenbaum Research Institute Study; German Lung Cancer Study and MD Anderson Cancer Center Study), which had already been analyzed separately with four different GSA methods (EASE; SLAT, mSUMSTAT and GenGen). This application revealed the pathway GO0015291 "transmembrane transporter activity" as significantly enriched with associated genes (GSA-method: EASE, p = 0.0315 corrected for multiple testing). Similar results were found for GO0015464 "acetylcholine receptor activity" but only when not corrected for multiple testing (all GSA-methods applied; p ≈ 0.02).
基因集分析(GSA)方法被用作全基因组关联研究(GWAS)的补充方法。对一组预定义基因的单标记关联估计值,要么与所有其余基因的估计值进行对比,要么与零假设的非关联背景进行对比。为了汇总来自多个基因集分析的p值,考虑单个标记关联点估计值在任何给定基因集上产生的观察模式的一致性非常重要。在此,我们提出了Fisher逆χ2方法META-GSA的增强版本,不过会对每项研究进行加权,以考虑关联模式之间的不完全相关性。
我们通过在20种不同情况下,对100个双等位基因标记处的500例病例和500例对照进行GWAS模拟来研究META-GSA的性能,在10个基因的基因集中模拟1到1.5之间的不同相对风险。将Wilcoxon秩和检验用作每项研究的基因集分析方法。我们发现,当假设10个基因中的5个基因的真实相对风险为1.5时,与简单汇总p值相比,META-GSA发现真正相关基因集的效能更高,例如分别为59%和37%。在感兴趣的基因集与其余基因集之间真实关联模式无差异的零假设下,两种方法的结果几乎不相关。我们建议在合并独立基因集分析的结果时,不要仅依赖p值。
我们应用META-GSA汇总四项肺癌风险病例对照GWAS(中欧研究和多伦多/伦嫩费尔德-塔嫩鲍姆研究所研究;德国肺癌研究和MD安德森癌症中心研究)的结果,这些研究之前已分别使用四种不同的基因集分析方法(EASE;SLAT、mSUMSTAT和GenGen)进行了分析。该应用揭示了通路GO0015291“跨膜转运蛋白活性”显著富集相关基因(基因集分析方法:EASE,经多重检验校正后p = 0.0315)。对于GO0015464“乙酰胆碱受体活性”也发现了类似结果,但仅在未进行多重检验校正时(应用所有基因集分析方法;p≈0.02)。