Pierson Emma, Yau Christopher
Department of Statistics, University of Oxford, 1 South Parks Road, OX1 3TG, Oxford, UK.
Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, OX3 7BN, Oxford, UK.
Genome Biol. 2015 Nov 2;16:241. doi: 10.1186/s13059-015-0805-z.
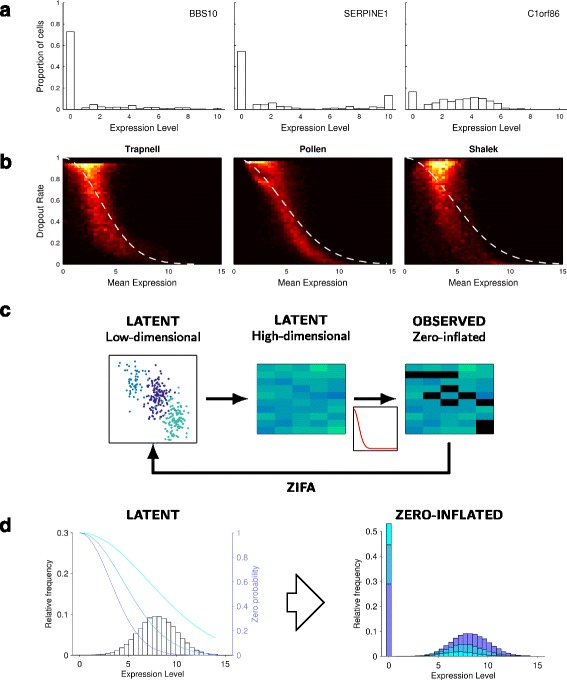
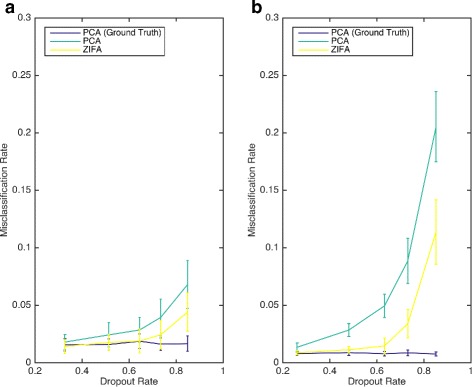
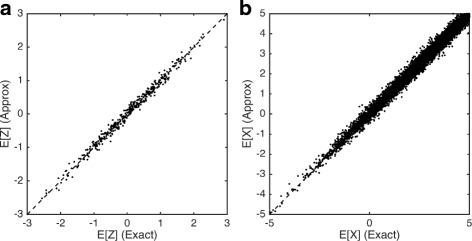
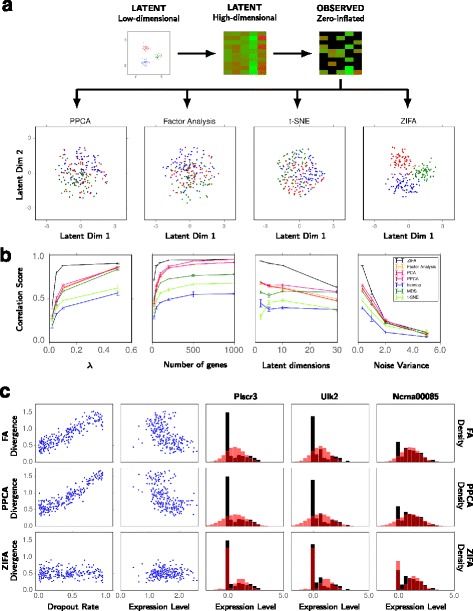
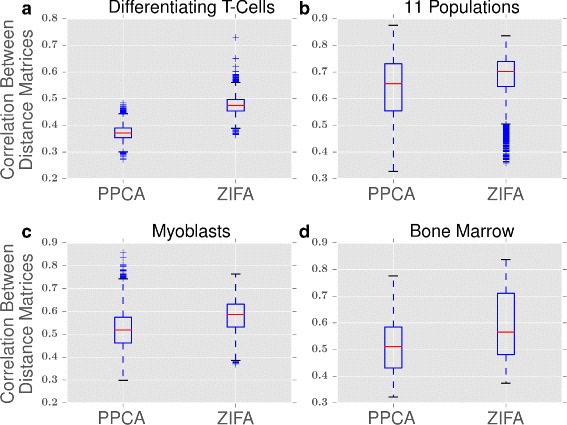
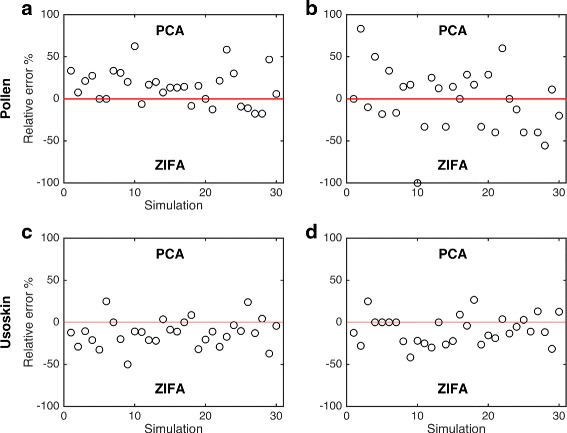
Single-cell RNA-seq data allows insight into normal cellular function and various disease states through molecular characterization of gene expression on the single cell level. Dimensionality reduction of such high-dimensional data sets is essential for visualization and analysis, but single-cell RNA-seq data are challenging for classical dimensionality-reduction methods because of the prevalence of dropout events, which lead to zero-inflated data. Here, we develop a dimensionality-reduction method, (Z)ero (I)nflated (F)actor (A)nalysis (ZIFA), which explicitly models the dropout characteristics, and show that it improves modeling accuracy on simulated and biological data sets.
单细胞RNA测序数据能够通过在单细胞水平上对基因表达进行分子特征分析,洞察正常细胞功能和各种疾病状态。对这类高维数据集进行降维对于可视化和分析至关重要,但由于缺失事件普遍存在,导致数据出现零膨胀,单细胞RNA测序数据对经典降维方法来说具有挑战性。在此,我们开发了一种降维方法,即零膨胀因子分析(ZIFA),该方法明确地对缺失特征进行建模,并表明它提高了对模拟数据集和生物数据集的建模准确性。