Lange Joanna, Wyrwicz Lucjan S, Vriend Gert
Laboratory of Bioinformatics and Biostatistics, M. Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw, Poland and Laboratory of Bioinformatics and Biostatistics, M. Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw, Poland and.
Laboratory of Bioinformatics and Biostatistics, M. Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw, Poland and.
Bioinformatics. 2016 Mar 15;32(6):932-6. doi: 10.1093/bioinformatics/btv663. Epub 2015 Nov 14.
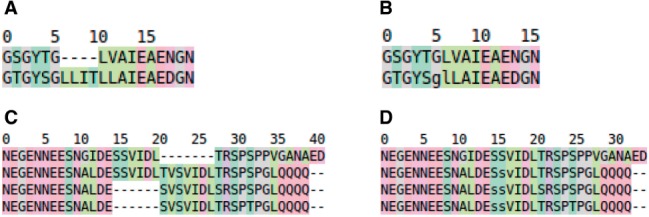
Intrinsically disordered proteins (IDPs) lack tertiary structure and thus differ from globular proteins in terms of their sequence-structure-function relations. IDPs have lower sequence conservation, different types of active sites and a different distribution of functionally important regions, which altogether make their multiple sequence alignment (MSA) difficult. The KMAD MSA software has been written specifically for the alignment and annotation of IDPs. It augments the substitution matrix with knowledge about post-translational modifications, functional domains and short linear motifs.
MSAs produced with KMAD describe well-conserved features among IDPs, tend to agree well with biological intuition, and are a good basis for designing new experiments to shed light on this large, understudied class of proteins.
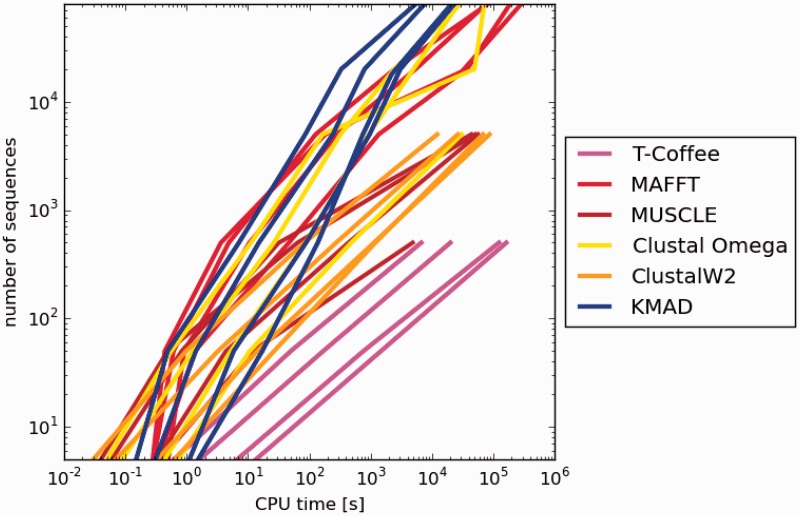
KMAD web server is accessible at http://www.cmbi.ru.nl/kmad/ A standalone version is freely available.
内在无序蛋白(IDP)缺乏三级结构,因此在序列-结构-功能关系方面与球状蛋白不同。IDP具有较低的序列保守性、不同类型的活性位点以及功能重要区域的不同分布,这些因素共同使得它们的多序列比对(MSA)变得困难。KMAD MSA软件是专门为IDP的比对和注释而编写的。它利用有关翻译后修饰、功能域和短线性基序的知识增强了替换矩阵。
用KMAD生成的MSA描述了IDP中保守良好的特征,往往与生物学直觉高度一致,并且是设计新实验以阐明这类大量研究不足的蛋白质的良好基础。
可通过http://www.cmbi.ru.nl/kmad/访问KMAD网络服务器。独立版本可免费获取。