Hizukuri Yoshiyuki, Sawada Ryusuke, Yamanishi Yoshihiro
Faculty of Exploratory Technology, Asubio Pharma Co. Ltd., 6-4-3 Minatojima-Minamimachi, Chuo-ku, Kobe, Hyogo, 650-0047, Japan.
Division of System Cohort, Multi-Scale Research Center for Medical Science, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka, 812-8582, Japan.
BMC Med Genomics. 2015 Dec 18;8:82. doi: 10.1186/s12920-015-0158-1.
Phenotype-based high-throughput screening is a useful technique for identifying drug candidate compounds that have a desired phenotype. However, the molecular mechanisms of the hit compounds remain unknown, and substantial effort is required to identify the target proteins associated with the phenotype.
In this study, we propose a new method to predict target proteins of drug candidate compounds based on drug-induced gene expression data in Connectivity Map and a machine learning classification technique, which we call the "transcriptomic approach."
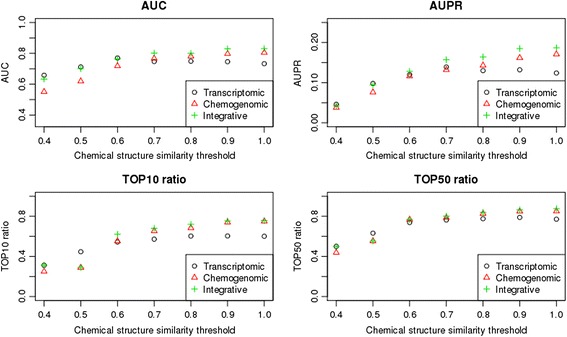
Unlike existing methods such as the chemogenomic approach, the transcriptomic approach enabled the prediction of target proteins without dependence on prior knowledge of compound chemical structures. The prediction accuracy of the chemogenomic approach was highly depended on compounds structure similarities in data sets. In contrast, the prediction accuracy of the transcriptomic approach was maintained at a sufficient level, even for benchmark data consisting of structurally diverse compounds.
The transcriptomic approach reported here is expected to be a useful tool for structure-independent prediction of target proteins for drug candidate compounds.
基于表型的高通量筛选是一种用于识别具有所需表型的候选药物化合物的有用技术。然而,命中化合物的分子机制仍然未知,并且需要大量努力来鉴定与该表型相关的靶蛋白。
在本研究中,我们提出了一种基于连通性图谱中的药物诱导基因表达数据和机器学习分类技术来预测候选药物化合物靶蛋白的新方法,我们将其称为“转录组学方法”。
与化学基因组学方法等现有方法不同,转录组学方法能够在不依赖化合物化学结构先验知识的情况下预测靶蛋白。化学基因组学方法的预测准确性高度依赖于数据集中化合物的结构相似性。相比之下,即使对于由结构多样的化合物组成的基准数据,转录组学方法的预测准确性也能保持在足够的水平。
本文报道的转录组学方法有望成为一种用于独立于结构预测候选药物化合物靶蛋白的有用工具。