Smith Jennifer A, Zagel Alicia L, Sun Yan V, Dolinoy Dana C, Bielak Lawrence F, Peyser Patricia A, Turner Stephen T, Mosley Thomas H, Kardia Sharon Lr
Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, Michigan USA.
Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, Michigan USA; Center for Health Statistics, Washington State Department of Health, Olympia, Washington USA.
Hereditary Genet. 2014 Dec;3(3). doi: 10.4172/2161-1041.1000137. Epub 2014 Oct 1.
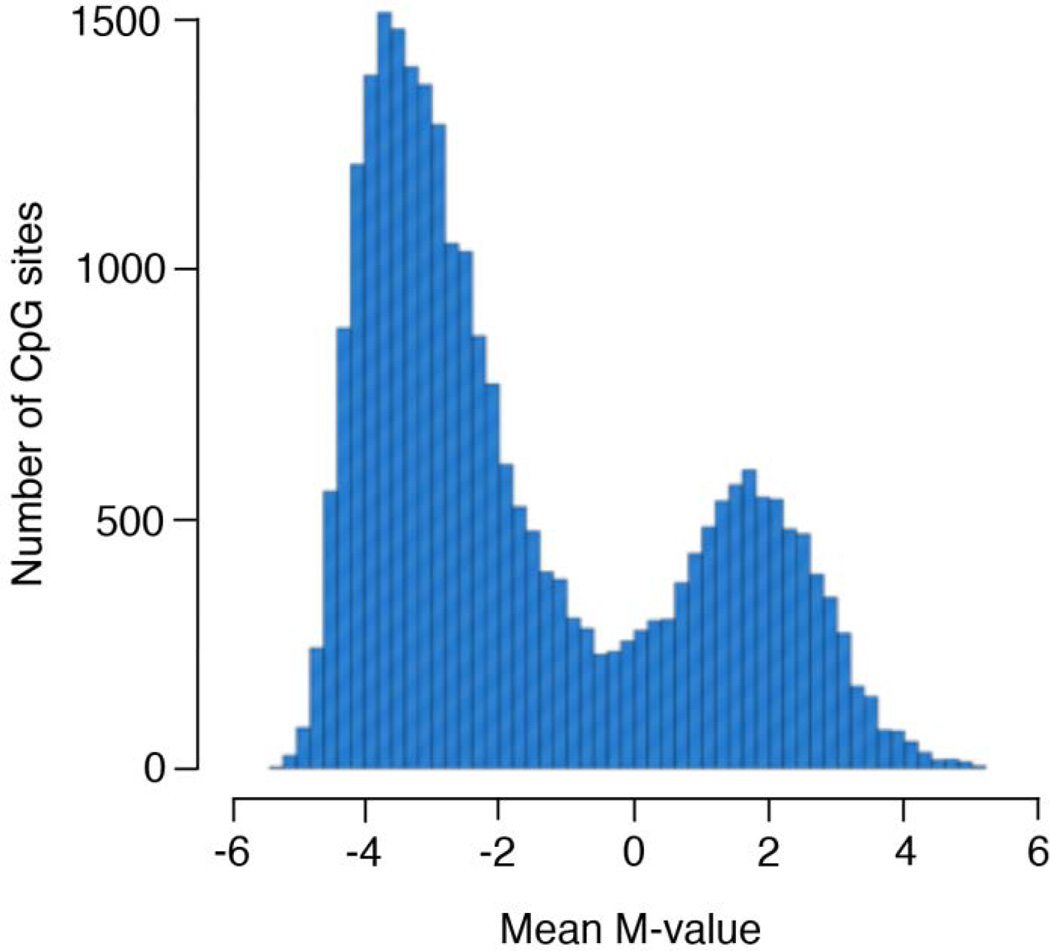
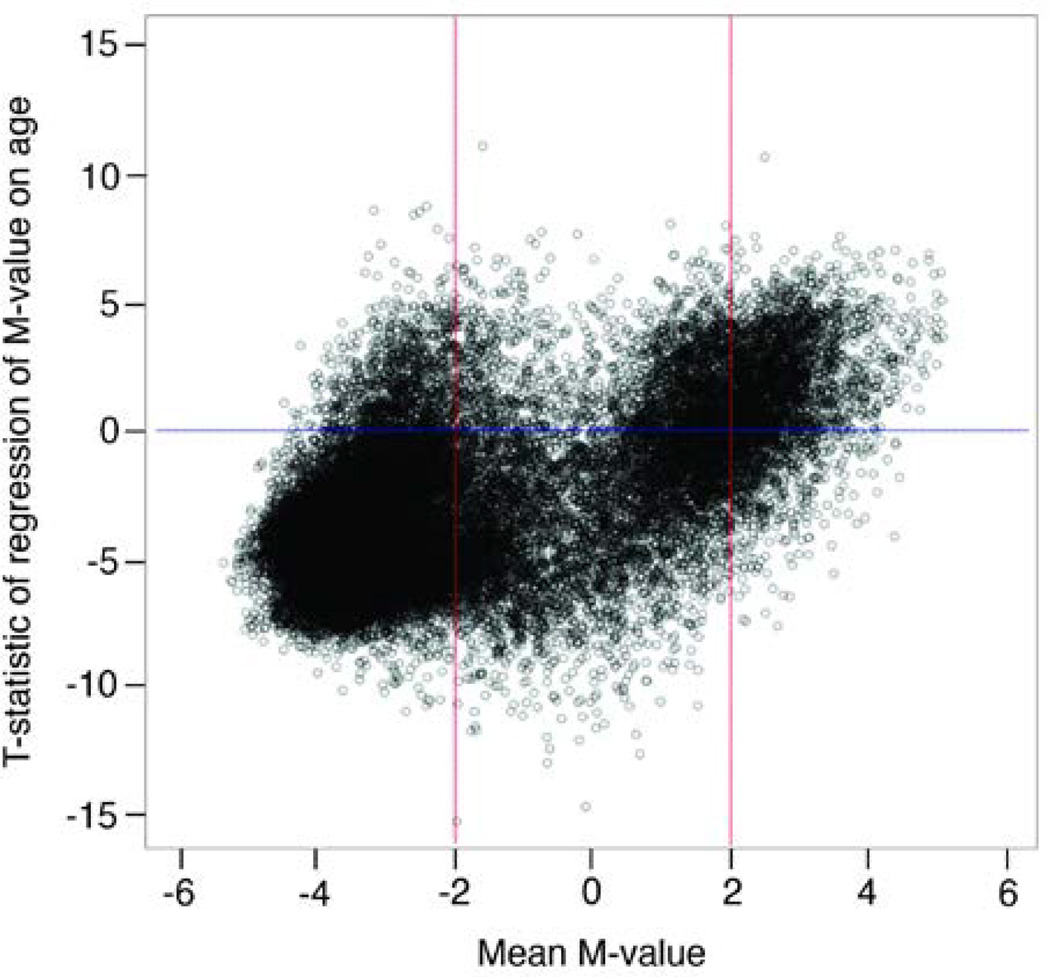
Age is a well-established risk factor for chronic diseases. However, the cellular and molecular changes associated with aging processes that are related to chronic disease initiation and progression are not well-understood. Thus, there is an increased need to identify new markers of cellular and molecular changes that occur during aging processes. In this study, we use genome-wide DNA methylation from 26,428 CpG sites in 13,877 genes to investigate the relationship between age and epigenetic variation in the peripheral blood cells of 972 African American adults from the Genetic Epidemiology Network of Arteriopathy (GENOA) study (mean age=66.3 years, range=39-95). Age was significantly associated with 7,601 (28.8%) CpG sites after Bonferroni correction for α=0.05 (<1.89×10). Due to the extraordinarily strong associations between age and many of the CpG sites (>7,000 sites with -values ranging from 10 to 10), we investigated how well the DNA methylation markers predict age. We found that 2,095 (7.9%) CpG sites were significant predictors of age after Bonferroni correction. The top five principal components of the 2,095 age-associated CpG sites accounted for 69.3% of the variability in these CpG sites, and they explained 26.8% of the variation in age. The associations between methylation markers and adult age are so ubiquitous and strong that we hypothesize that DNA methylation patterns may be an important measure of cellular aging processes. Given the highly correlated nature of the age-associated epigenome (as evidenced by the principal components analysis), whole pathways may be regulated as a consequence of aging.
年龄是一种公认的慢性疾病风险因素。然而,与衰老过程相关的、与慢性疾病发生和发展有关的细胞和分子变化尚未得到充分理解。因此,越来越需要确定衰老过程中发生的细胞和分子变化的新标志物。在本研究中,我们使用来自13877个基因中26428个CpG位点的全基因组DNA甲基化,来研究年龄与动脉病遗传流行病学网络(GENOA)研究中972名非裔美国成年人外周血细胞中表观遗传变异之间的关系(平均年龄 = 66.3岁,范围 = 39 - 95岁)。在对α = 0.05进行Bonferroni校正后(<1.89×10),年龄与7601个(28.8%)CpG位点显著相关。由于年龄与许多CpG位点之间存在极强的关联(超过7000个位点,P值范围从10到10),我们研究了DNA甲基化标志物预测年龄的能力。我们发现,在进行Bonferroni校正后,2095个(7.9%)CpG位点是年龄的显著预测因子。2095个与年龄相关的CpG位点的前五个主成分占这些CpG位点变异性的69.3%,它们解释了年龄变异的26.8%。甲基化标志物与成年人年龄之间的关联如此普遍且强烈,以至于我们推测DNA甲基化模式可能是细胞衰老过程的一个重要指标。鉴于与年龄相关的表观基因组具有高度相关性(主成分分析证明了这一点),整个通路可能会因衰老而受到调控。