Williams Claire R, Baccarella Alyssa, Parrish Jay Z, Kim Charles C
Department of Biology, University of Washington, Seattle, WA, 98195, USA.
Division of Experimental Medicine, Department of Medicine, University of California San Francisco, San Francisco, CA, 94110, USA.
BMC Bioinformatics. 2016 Feb 25;17:103. doi: 10.1186/s12859-016-0956-2.
High-throughput RNA-Sequencing (RNA-Seq) has become the preferred technique for studying gene expression differences between biological samples and for discovering novel isoforms, though the techniques to analyze the resulting data are still immature. One pre-processing step that is widely but heterogeneously applied is trimming, in which low quality bases, identified by the probability that they are called incorrectly, are removed. However, the impact of trimming on subsequent alignment to a genome could influence downstream analyses including gene expression estimation; we hypothesized that this might occur in an inconsistent manner across different genes, resulting in differential bias.
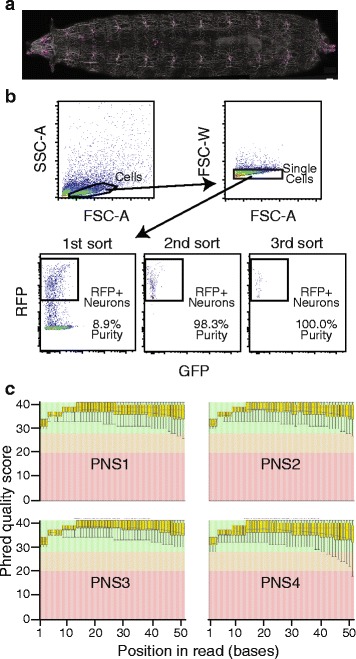
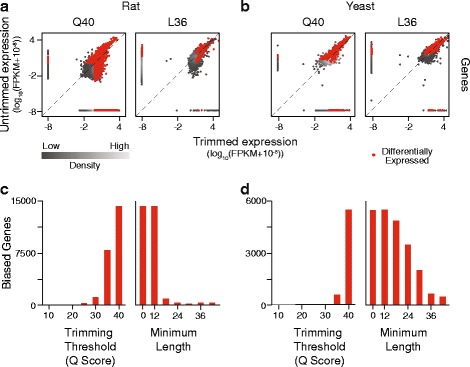
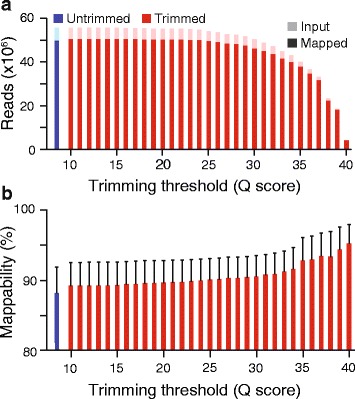
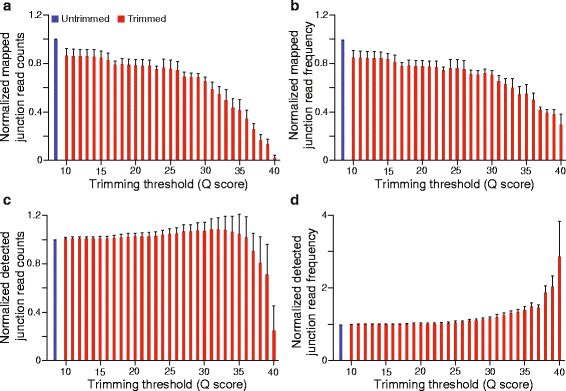
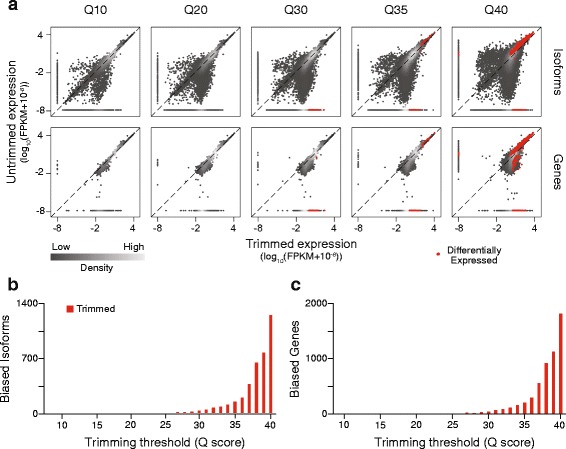
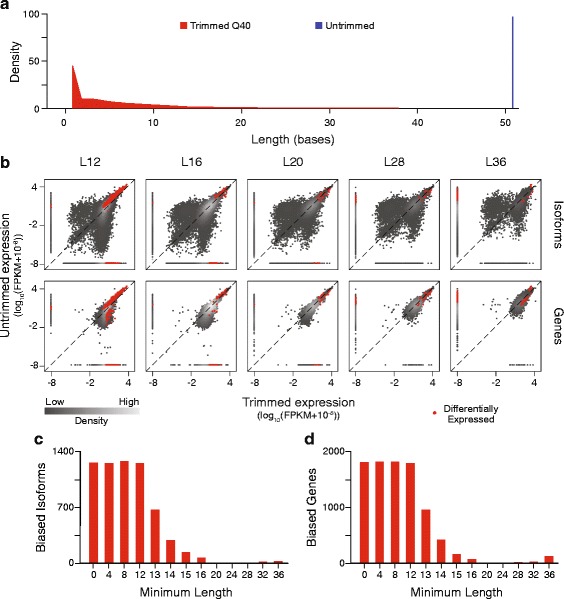
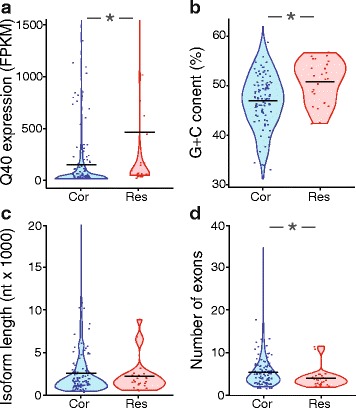
To assess the effects of trimming on gene expression, we generated RNA-Seq data sets from four samples of larval Drosophila melanogaster sensory neurons, and used three trimming algorithms--SolexaQA, Trimmomatic, and ConDeTri-to perform quality-based trimming across a wide range of stringencies. After aligning the reads to the D. melanogaster genome with TopHat2, we used Cuffdiff2 to compare the original, untrimmed gene expression estimates to those following trimming. With the most aggressive trimming parameters, over ten percent of genes had significant changes in their estimated expression levels. This trend was seen with two additional RNA-Seq data sets and with alternative differential expression analysis pipelines. We found that the majority of the expression changes could be mitigated by imposing a minimum length filter following trimming, suggesting that the differential gene expression was primarily being driven by spurious mapping of short reads. Slight differences with the untrimmed data set remained after length filtering, which were associated with genes with low exon numbers and high GC content. Finally, an analysis of paired RNA-seq/microarray data sets suggests that no or modest trimming results in the most biologically accurate gene expression estimates.
We find that aggressive quality-based trimming has a large impact on the apparent makeup of RNA-Seq-based gene expression estimates, and that short reads can have a particularly strong impact. We conclude that implementation of trimming in RNA-Seq analysis workflows warrants caution, and if used, should be used in conjunction with a minimum read length filter to minimize the introduction of unpredictable changes in expression estimates.
高通量RNA测序(RNA-Seq)已成为研究生物样本间基因表达差异以及发现新异构体的首选技术,不过分析所得数据的技术仍不成熟。一个广泛但应用方式各异的预处理步骤是修剪,即通过碱基被错误识别的概率来确定低质量碱基并将其去除。然而,修剪对后续与基因组比对的影响可能会影响包括基因表达估计在内的下游分析;我们推测这种情况在不同基因间可能以不一致的方式发生,从而导致差异偏差。
为评估修剪对基因表达的影响,我们从黑腹果蝇幼虫感觉神经元的四个样本中生成了RNA-Seq数据集,并使用三种修剪算法——SolexaQA、Trimmomatic和ConDeTri——在广泛的严格度范围内进行基于质量的修剪。在用TopHat2将 reads 比对到黑腹果蝇基因组后,我们使用Cuffdiff2比较原始的、未修剪的基因表达估计值与修剪后的估计值。使用最激进的修剪参数时,超过10%的基因在其估计表达水平上有显著变化。在另外两个RNA-Seq数据集以及替代的差异表达分析流程中也观察到了这种趋势。我们发现,通过在修剪后施加最小长度过滤,大多数表达变化可以得到缓解,这表明差异基因表达主要是由短 reads 的错误比对驱动的。长度过滤后,与未修剪数据集仍存在细微差异,这些差异与外显子数量少和GC含量高的基因有关。最后,对配对的RNA-seq/微阵列数据集的分析表明,不进行修剪或适度修剪会得到最符合生物学实际的基因表达估计值。
我们发现,基于质量的激进修剪对基于RNA-Seq的基因表达估计的表观组成有很大影响,并且短 reads 可能有特别强烈的影响。我们得出结论,在RNA-Seq分析工作流程中实施修剪需要谨慎,如果使用,应与最小读长过滤结合使用,以尽量减少引入表达估计中不可预测的变化。