Speed Doug, Balding David J
UCL Genetics Institute, University College London, London WC1E 6BT, United Kingdom
UCL Genetics Institute, University College London, London WC1E 6BT, United Kingdom.
Genome Res. 2014 Sep;24(9):1550-7. doi: 10.1101/gr.169375.113. Epub 2014 Jun 24.
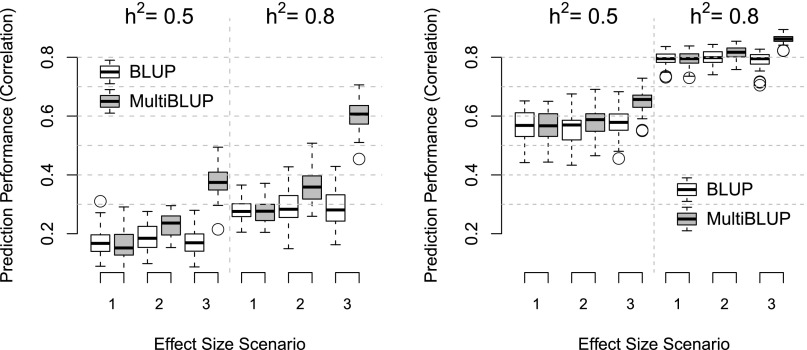
BLUP (best linear unbiased prediction) is widely used to predict complex traits in plant and animal breeding, and increasingly in human genetics. The BLUP mathematical model, which consists of a single random effect term, was adequate when kinships were measured from pedigrees. However, when genome-wide SNPs are used to measure kinships, the BLUP model implicitly assumes that all SNPs have the same effect-size distribution, which is a severe and unnecessary limitation. We propose MultiBLUP, which extends the BLUP model to include multiple random effects, allowing greatly improved prediction when the random effects correspond to classes of SNPs with distinct effect-size variances. The SNP classes can be specified in advance, for example, based on SNP functional annotations, and we also provide an adaptive procedure for determining a suitable partition of SNPs. We apply MultiBLUP to genome-wide association data from the Wellcome Trust Case Control Consortium (seven diseases), and from much larger studies of celiac disease and inflammatory bowel disease, finding that it consistently provides better prediction than alternative methods. Moreover, MultiBLUP is computationally very efficient; for the largest data set, which includes 12,678 individuals and 1.5 M SNPs, the total analysis can be run on a single desktop PC in less than a day and can be parallelized to run even faster. Tools to perform MultiBLUP are freely available in our software LDAK.
最佳线性无偏预测(BLUP)在动植物育种中被广泛用于预测复杂性状,在人类遗传学中的应用也日益增多。当通过系谱测量亲缘关系时,由单个随机效应项组成的BLUP数学模型是适用的。然而,当使用全基因组单核苷酸多态性(SNP)来测量亲缘关系时,BLUP模型隐含地假设所有SNP具有相同的效应大小分布,这是一个严格且不必要的限制。我们提出了多BLUP(MultiBLUP),它扩展了BLUP模型以包含多个随机效应,当随机效应对应于具有不同效应大小方差的SNP类别时,能大大提高预测效果。SNP类别可以提前指定,例如基于SNP功能注释,并且我们还提供了一种自适应程序来确定合适的SNP划分。我们将多BLUP应用于来自威康信托病例对照协会(七种疾病)的全基因组关联数据,以及来自乳糜泻和炎症性肠病的更大规模研究的数据,发现它始终比其他方法提供更好的预测。此外,多BLUP在计算上非常高效;对于包含12,678个个体和150万个SNP的最大数据集,整个分析可以在一台台式电脑上不到一天的时间内运行完成,并且可以并行运行以更快完成。执行多BLUP的工具可在我们的软件LDAK中免费获得。