Zhu Zheng, Chan Jasper Fuk-Woo, Tee Kah-Meng, Choi Garnet Kwan-Yue, Lau Susanna Kar-Pui, Woo Patrick Chiu-Yat, Tse Herman, Yuen Kwok-Yung
Department of Microbiology, The University of Hong Kong, Hong Kong, China.
State Key Laboratory of Emerging Infectious Diseases, The University of Hong Kong, Hong Kong, China.
Emerg Microbes Infect. 2016 Mar 16;5(3):e22. doi: 10.1038/emi.2016.48.
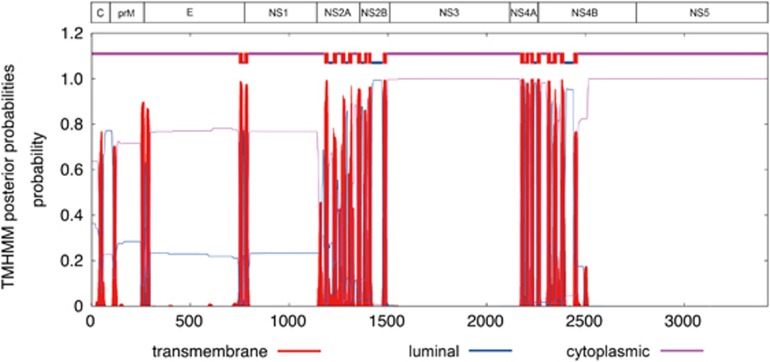
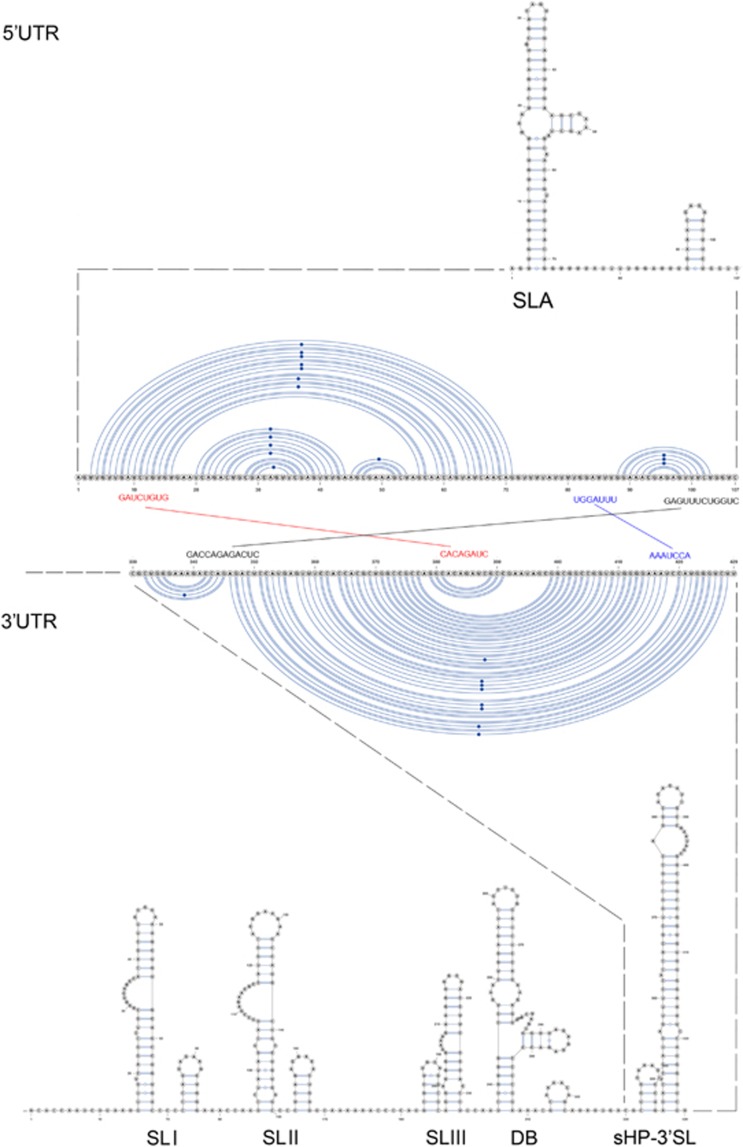
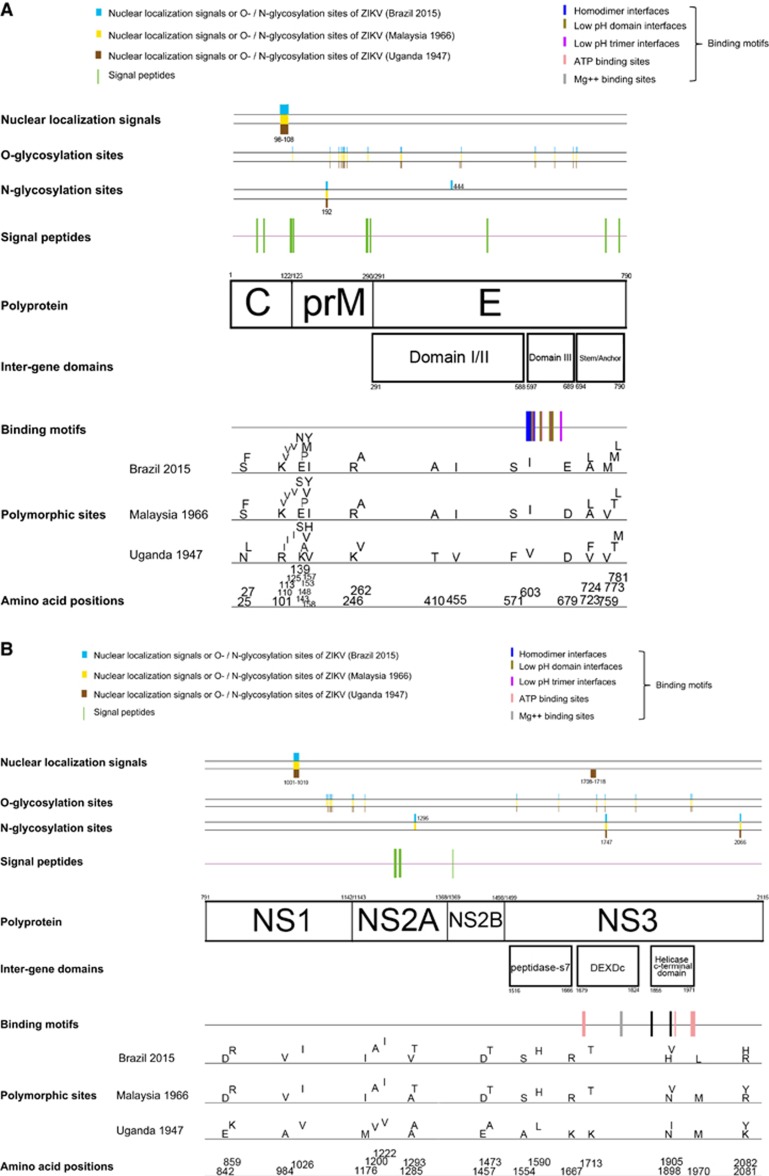
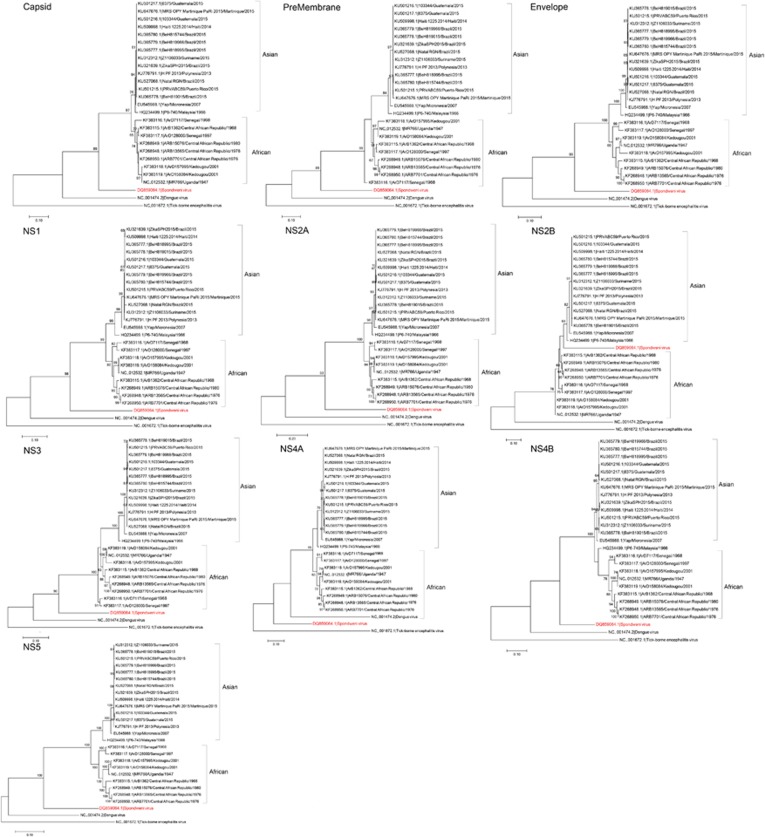
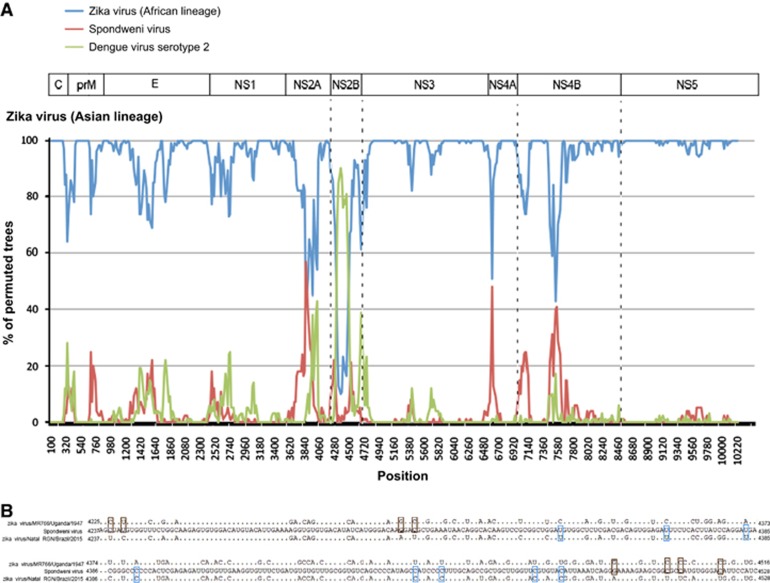
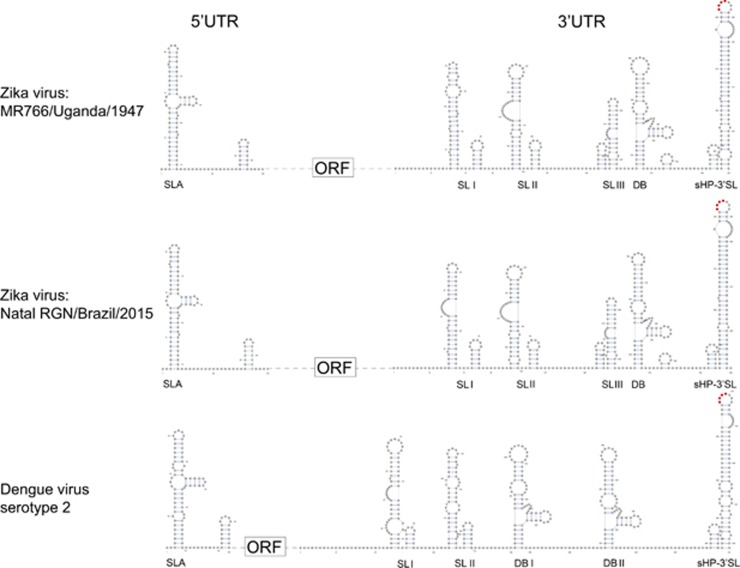
Less than 20 sporadic cases of human Zika virus (ZIKV) infection were reported in Africa and Asia before 2007, but large outbreaks involving up to 73% of the populations on the Pacific islands have started since 2007, and spread to the Americas in 2014. Moreover, the clinical manifestation of ZIKV infection has apparently changed, as evident by increasing reports of neurological complications, such as Guillain-Barré syndrome in adults and congenital anomalies in neonates. We comprehensively compared the genome sequences of pre-epidemic and epidemic ZIKV strains with complete genome or complete polyprotein sequences available in GenBank. Besides the reported phylogenetic clustering of the epidemic strains with the Asian lineage, we found that the topology of phylogenetic tree of all coding regions is the same except that of the non-structural 2B (NS2B) coding region. This finding was confirmed by bootscan analysis and multiple sequence alignment, which suggested the presence of a fragment of genetic recombination at NS2B with that of Spondweni virus. Moreover, the representative epidemic strain possesses one large bulge of nine bases instead of an external loop on the first stem-loop structure at the 3'-untranslated region just distal to the stop codon of the NS5 in the 1947 pre-epidemic prototype strain. Fifteen amino acid substitutions are found in the epidemic strains when compared with the pre-epidemic strains. As mutations in other flaviviruses can be associated with changes in virulence, replication efficiency, antigenic epitopes and host tropism, further studies would be important to ascertain the biological significance of these genomic changes.
2007年之前,非洲和亚洲报告的人类寨卡病毒(ZIKV)感染散发病例不足20例,但自2007年以来,太平洋岛屿上爆发了大规模疫情,波及多达73%的人口,并于2014年蔓延至美洲。此外,ZIKV感染的临床表现显然已经发生变化,成人吉兰-巴雷综合征和新生儿先天性异常等神经并发症的报告增多就证明了这一点。我们全面比较了流行前和流行期间ZIKV毒株的基因组序列,这些序列的完整基因组或完整多聚蛋白序列可在GenBank中获取。除了报告的流行毒株与亚洲谱系的系统发育聚类外,我们发现所有编码区的系统发育树拓扑结构相同,除了非结构2B(NS2B)编码区。这一发现通过bootscan分析和多序列比对得到证实,这表明NS2B存在与斯庞德温尼病毒的基因重组片段。此外,在1947年流行前的原型毒株中,代表流行毒株在NS5终止密码子远端的3'非翻译区的第一个茎环结构上有一个由九个碱基组成的大凸起,而不是外部环。与流行前毒株相比,流行毒株中发现了15个氨基酸替换。由于其他黄病毒的突变可能与毒力、复制效率、抗原表位和宿主嗜性的变化有关,进一步的研究对于确定这些基因组变化的生物学意义很重要。