Parida S, Pal I, Parekh A, Thakur B, Bharti R, Das S, Mandal M
School of Medical Science and Technology, Indian Institute of Technology, Kharagpur, West Bengal 721302, India.
National Institute of Cholera and Enteric Diseases, P-33, C.I.T. Road, Scheme XM, Beleghata, Kolkata, West Bengal 700010, India.
Cell Death Dis. 2016 Mar 24;7(3):e2154. doi: 10.1038/cddis.2016.61.
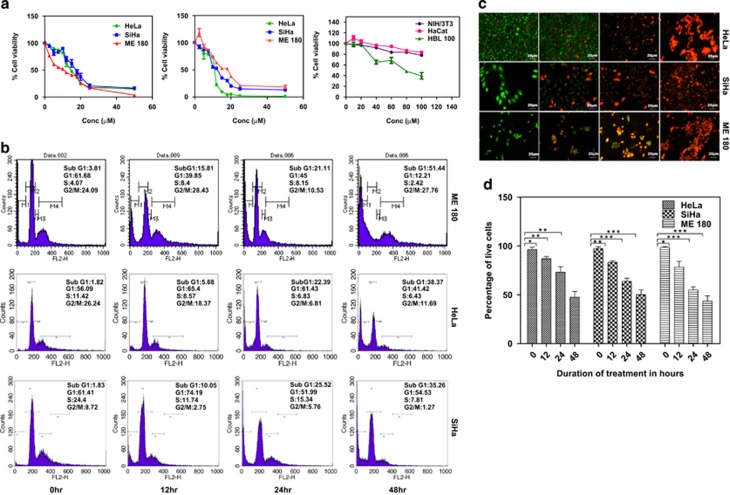
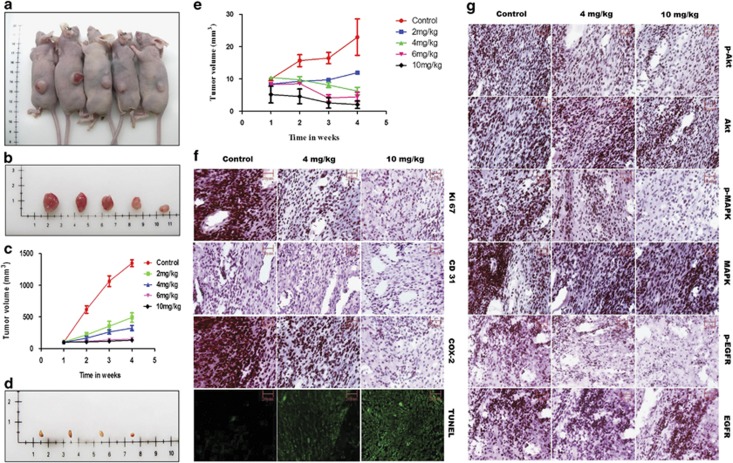
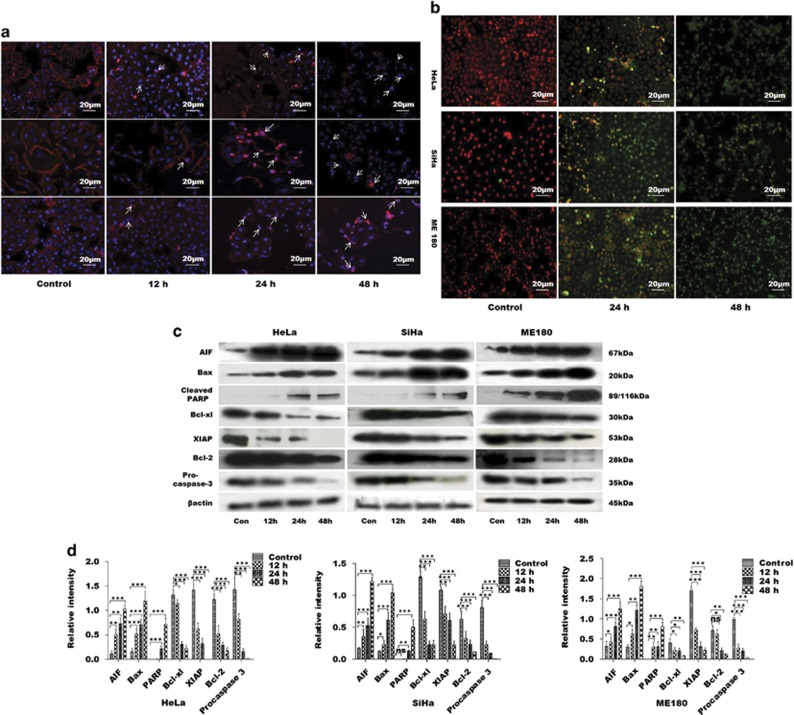
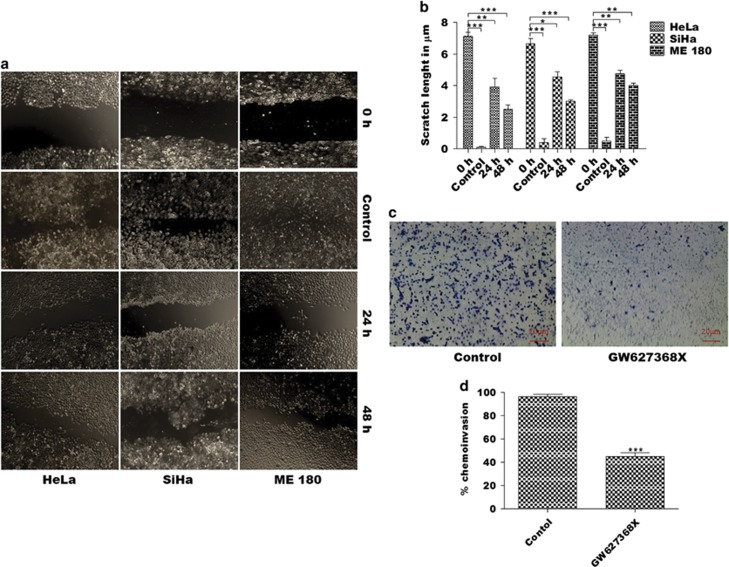
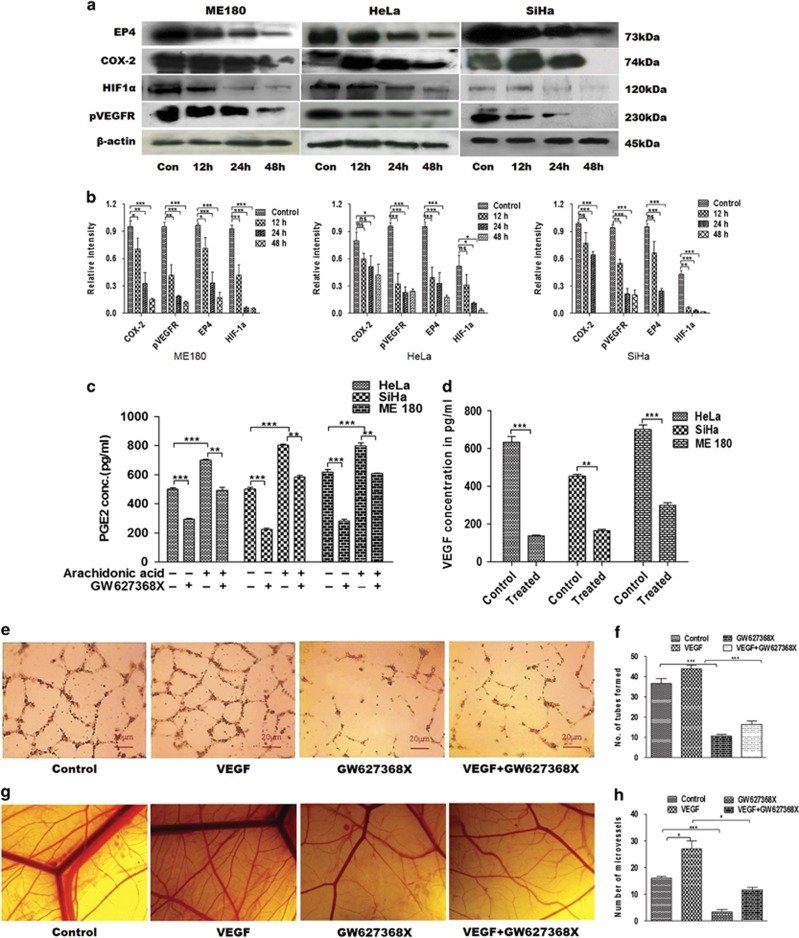
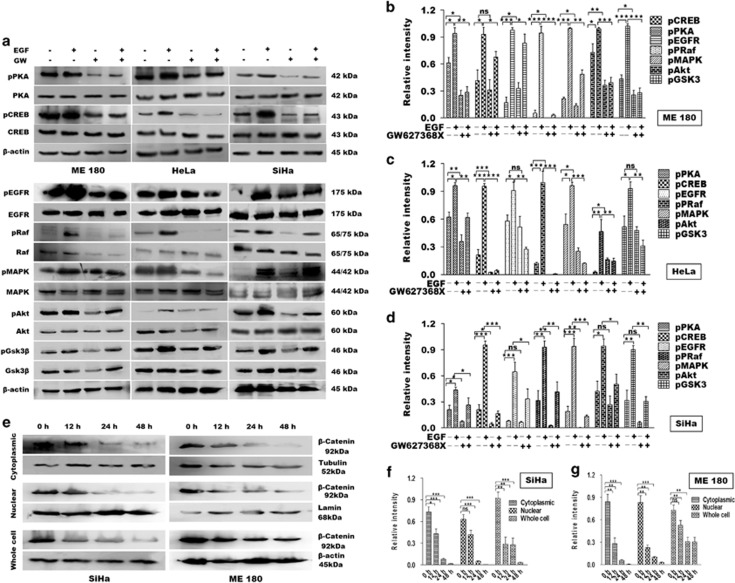
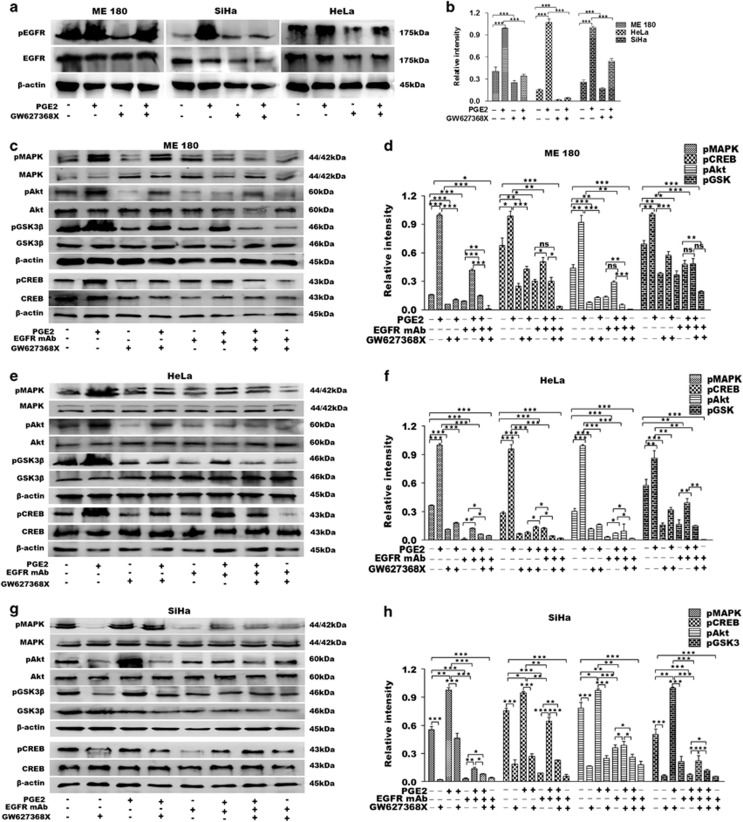
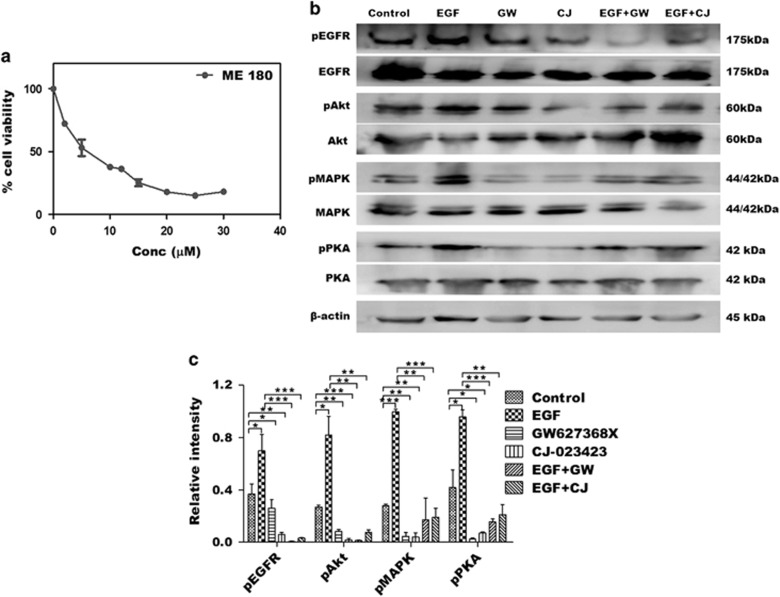
PGE2, the major product of cyclooxygenases implicated in carcinogenesis, is significantly upregulated in cervical cancer. PGE2 via prostanoid receptor EP4 stimulates proliferation and motility while inhibiting apoptosis and immune surveillance. It promotes angiogenesis by stimulating the production of pro-angiogenic factors. The present study demonstrates GW627368X, a highly selective competitive EP4 antagonist, which hinders cervical cancer progression by inhibiting EP4/epithelial growth factor receptor (EGFR) interactive signaling. GW627368X reduced protein kinase A (PKA) phosphorylation which in turn leads to decreased cAMP response element-binding protein (CREB) activation. Decreased PKA phosphorylation also directly enhanced Bax activity and in part reduced glycogen synthase kinase 3 (GSK3)β phosphorylation. Owing to the interactive signaling between EP4 and EGFR, GW627368X lowered EGFR phosphorylation in turn reducing Akt, mitogen-activated protein kinase (MAPK) and GSK3β activity significantly. Sublethal dose of GW627368X was found to reduce the nuclear translocation of β-catenin in a time dependent manner along with time-dependent decrease in cytoplasmic as well as whole-cell β-catenin. Decreased CREB and β-catenin transcriptional activity restricts the aberrant transcription of key genes like EP4, cyclooxygenase (COX)-2, vascular endothelial growth factor and c-myc, which ultimately control cell survival, proliferation and angiogenesis. Reduced activity of EGFR resulted in enhanced expression of 15-hydroxyprostaglandin dehydrogenase increasing PGE2 degradation thereby blocking a positive feedback loop. In xenograft model, dose-dependent decrease in cancer proliferation was observed characterized by reduction in tumor mass and volume and a marked decrease in Ki67 expression. A diminished CD31 specific staining signified decreased tumor angiogenesis. Reduced expression of pAkt, pMAPK, pEGFR and COX-2 validated in vitro results. GW627368X therefore effectively inhibits tumor survival, motility, proliferation and angiogenesis by blocking EP4/EGFR interactive signaling. EP4 is a potent therapeutic target in cervical cancer and can be explored in combination with conventional therapies to attain superior outcomes and to overcome complications associated with organ toxicities, therapeutic resistance and disease relapse.
前列腺素E2(PGE2)是参与致癌作用的环氧化酶的主要产物,在宫颈癌中显著上调。PGE2通过前列腺素受体EP4刺激增殖和迁移,同时抑制细胞凋亡和免疫监视。它通过刺激促血管生成因子的产生促进血管生成。本研究证明了GW627368X,一种高度选择性竞争性EP4拮抗剂,其通过抑制EP4/表皮生长因子受体(EGFR)相互作用信号传导来阻碍宫颈癌进展。GW627368X降低蛋白激酶A(PKA)磷酸化,进而导致环磷酸腺苷反应元件结合蛋白(CREB)激活减少。PKA磷酸化降低还直接增强了Bax活性,并部分降低了糖原合酶激酶3(GSK3)β磷酸化。由于EP4与EGFR之间的相互作用信号传导,GW627368X降低了EGFR磷酸化,进而显著降低Akt、丝裂原活化蛋白激酶(MAPK)和GSK3β活性。发现亚致死剂量的GW627368X以时间依赖性方式减少β-连环蛋白的核转位,同时细胞质以及全细胞β-连环蛋白随时间依赖性减少。CREB和β-连环蛋白转录活性降低限制了EP4、环氧化酶(COX)-2、血管内皮生长因子和c-myc等关键基因的异常转录,这些基因最终控制细胞存活、增殖和血管生成。EGFR活性降低导致15-羟基前列腺素脱氢酶表达增加,从而增加PGE2降解,从而阻断正反馈回路。在异种移植模型中,观察到癌症增殖呈剂量依赖性降低,表现为肿瘤质量和体积减小以及Ki67表达显著降低。CD31特异性染色减弱表明肿瘤血管生成减少。pAkt、pMAPK、pEGFR和COX-2表达降低验证了体外实验结果。因此,GW627368X通过阻断EP4/EGFR相互作用信号传导有效抑制肿瘤存活、迁移、增殖和血管生成。EP4是宫颈癌中一个有效的治疗靶点,可以与传统疗法联合探索,以获得更好的疗效,并克服与器官毒性、治疗耐药性和疾病复发相关的并发症。