Swift Robert V, Jusoh Siti A, Offutt Tavina L, Li Eric S, Amaro Rommie E
Department of Chemistry and Biochemistry, University of California, San Diego , La Jolla, California 92093-0340, United States.
Faculty of Pharmacy, Universiti Teknologi MARA , 42300 Bandar Puncak Alam, Malaysia.
J Chem Inf Model. 2016 May 23;56(5):830-42. doi: 10.1021/acs.jcim.5b00684. Epub 2016 May 3.
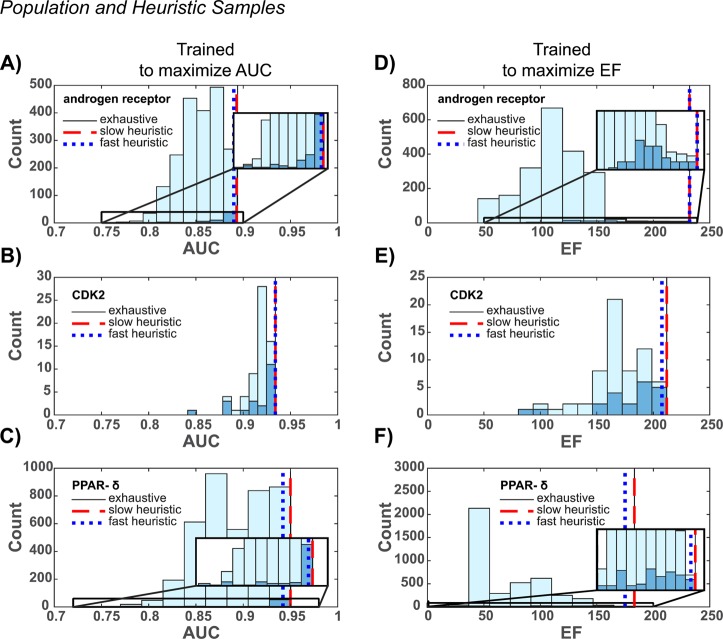
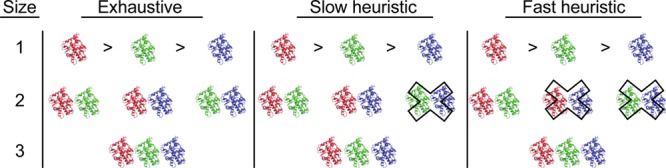
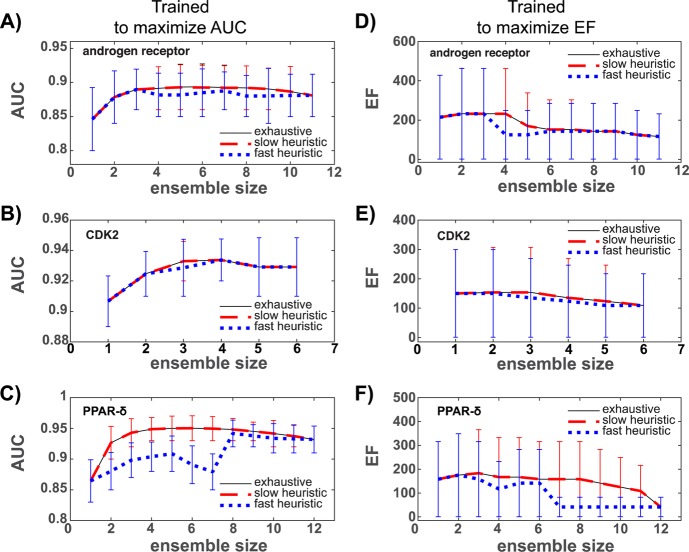
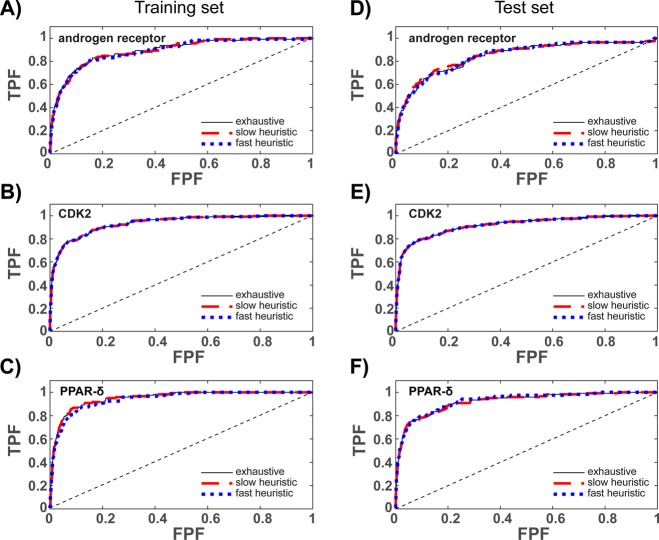
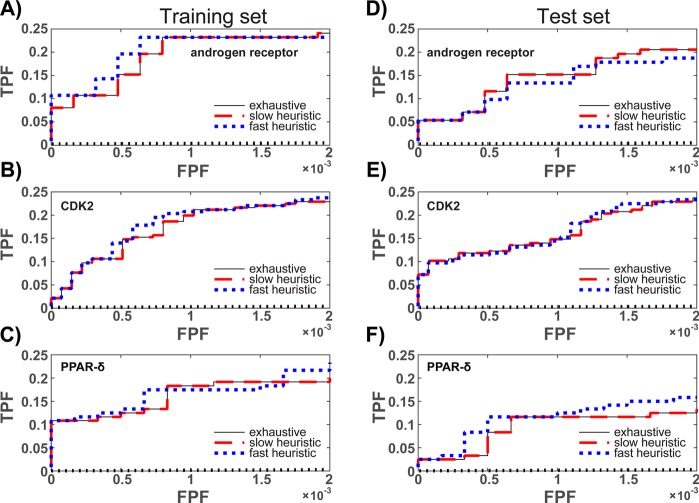
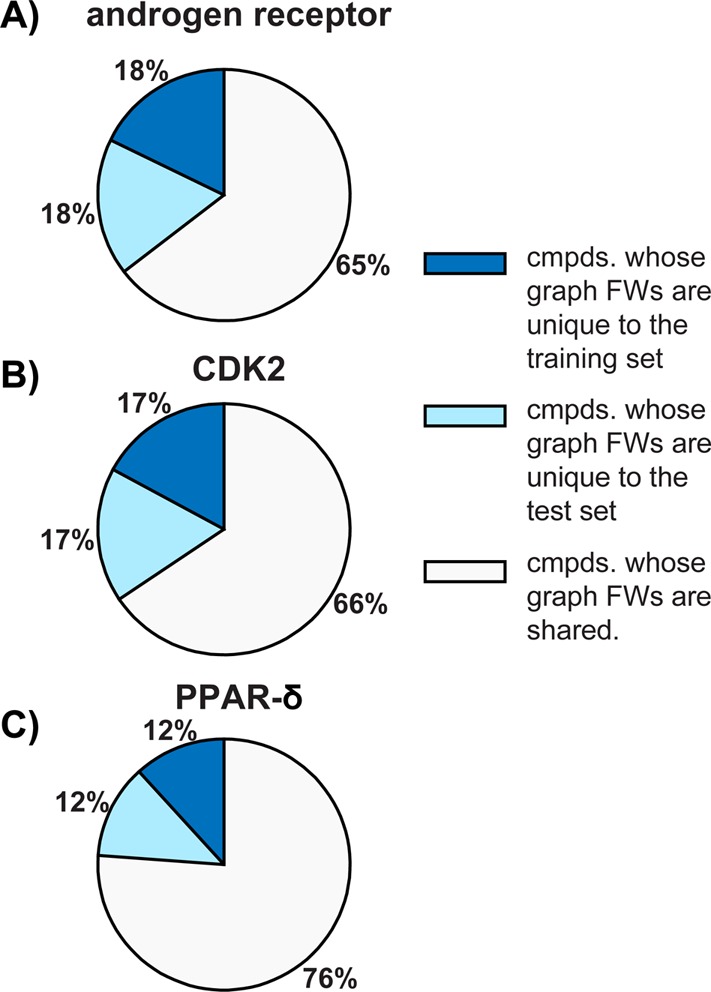
Ensemble docking can be a successful virtual screening technique that addresses the innate conformational heterogeneity of macromolecular drug targets. Yet, lacking a method to identify a subset of conformational states that effectively segregates active and inactive small molecules, ensemble docking may result in the recommendation of a large number of false positives. Here, three knowledge-based methods that construct structural ensembles for virtual screening are presented. Each method selects ensembles by optimizing an objective function calculated using the receiver operating characteristic (ROC) curve: either the area under the ROC curve (AUC) or a ROC enrichment factor (EF). As the number of receptor conformations, N, becomes large, the methods differ in their asymptotic scaling. Given a set of small molecules with known activities and a collection of target conformations, the most resource intense method is guaranteed to find the optimal ensemble but scales as O(2(N)). A recursive approximation to the optimal solution scales as O(N(2)), and a more severe approximation leads to a faster method that scales linearly, O(N). The techniques are generally applicable to any system, and we demonstrate their effectiveness on the androgen nuclear hormone receptor (AR), cyclin-dependent kinase 2 (CDK2), and the peroxisome proliferator-activated receptor δ (PPAR-δ) drug targets. Conformations that consisted of a crystal structure and molecular dynamics simulation cluster centroids were used to form AR and CDK2 ensembles. Multiple available crystal structures were used to form PPAR-δ ensembles. For each target, we show that the three methods perform similarly to one another on both the training and test sets.
整合对接可以是一种成功的虚拟筛选技术,可解决大分子药物靶点固有的构象异质性问题。然而,由于缺乏一种识别构象状态子集的方法,而该子集能有效区分活性和非活性小分子,整合对接可能会导致推荐大量假阳性结果。在此,我们提出了三种基于知识的方法,用于构建用于虚拟筛选的结构集合。每种方法通过优化使用接收器操作特征(ROC)曲线计算的目标函数来选择集合:即ROC曲线下面积(AUC)或ROC富集因子(EF)。随着受体构象数量N变得很大,这些方法在渐近缩放方面存在差异。给定一组具有已知活性的小分子和一组目标构象,资源消耗最大的方法保证能找到最优集合,但缩放比例为O(2(N))。最优解的递归近似缩放比例为O(N(2)),更粗略的近似会得到一种缩放比例为线性O(N)的更快方法。这些技术通常适用于任何系统,我们在雄激素核激素受体(AR)、细胞周期蛋白依赖性激酶2(CDK2)和过氧化物酶体增殖物激活受体δ(PPAR-δ)药物靶点上证明了它们的有效性。由晶体结构和分子动力学模拟聚类中心组成的构象用于形成AR和CDK2集合。使用多个可用的晶体结构来形成PPAR-δ集合。对于每个靶点,我们表明这三种方法在训练集和测试集上的表现彼此相似。