Zhou Xiang, Li Rui, Michal Jennifer J, Wu Xiao-Lin, Liu Zhongzhen, Zhao Hui, Xia Yin, Du Weiwei, Wildung Mark R, Pouchnik Derek J, Harland Richard M, Jiang Zhihua
Department of Animal Sciences and Center for Reproductive Biology, Washington State University, Pullman, Washington 99164-7620.
School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong.
Genetics. 2016 Jun;203(2):683-97. doi: 10.1534/genetics.116.188508. Epub 2016 Apr 20.
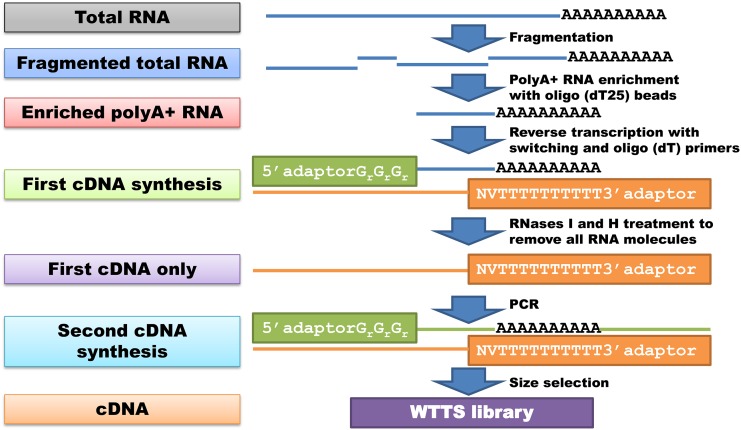
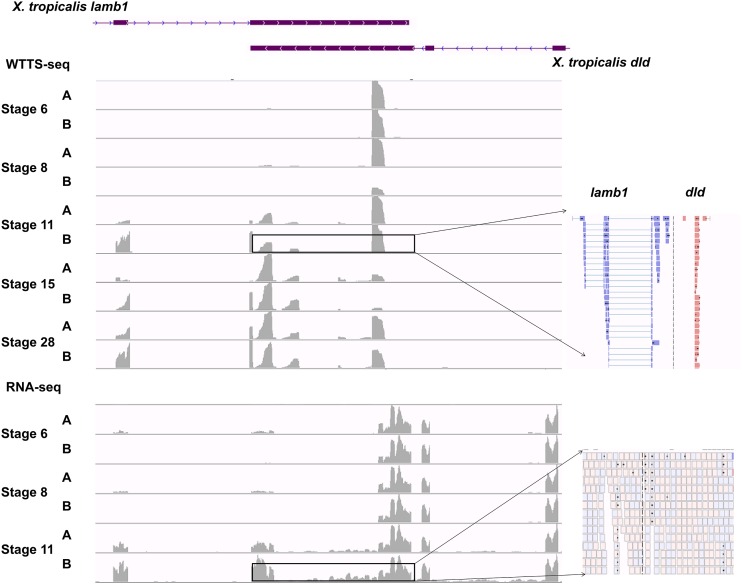
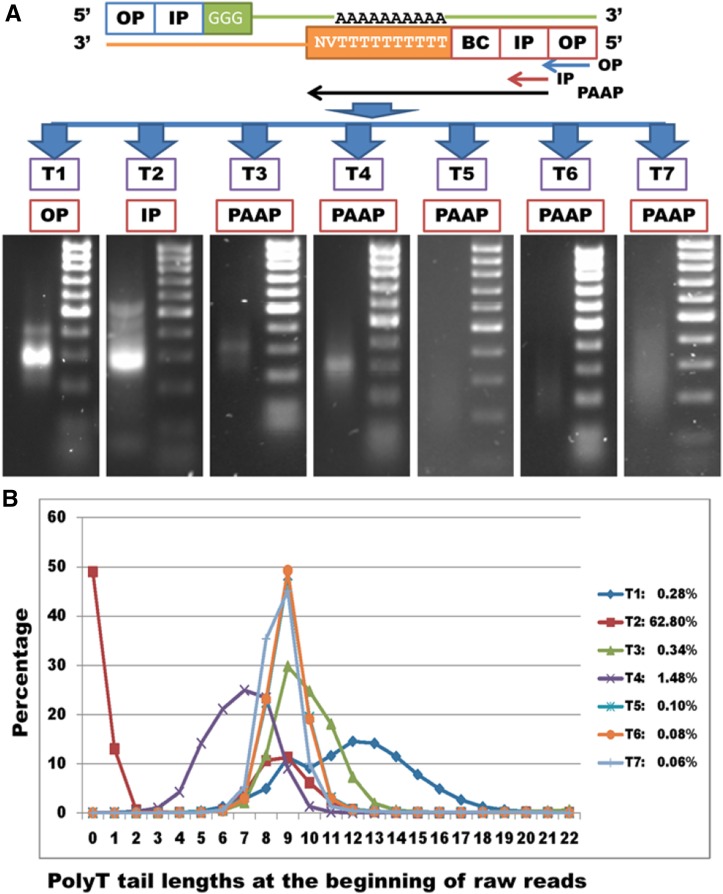
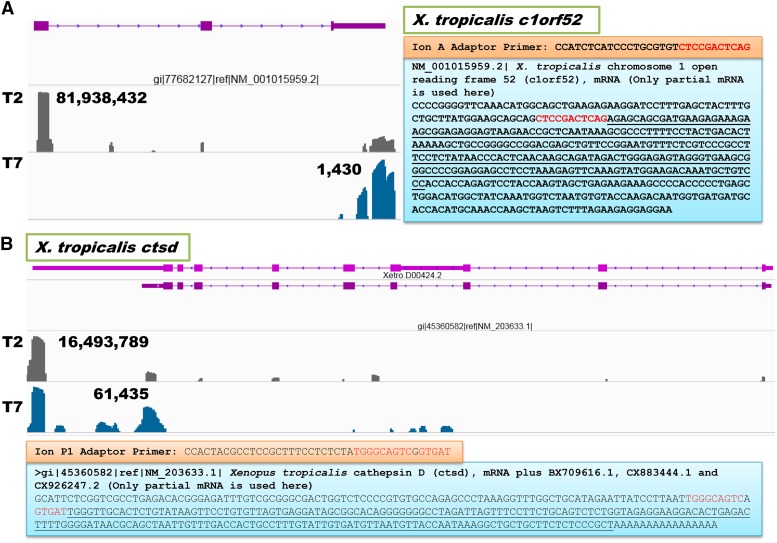
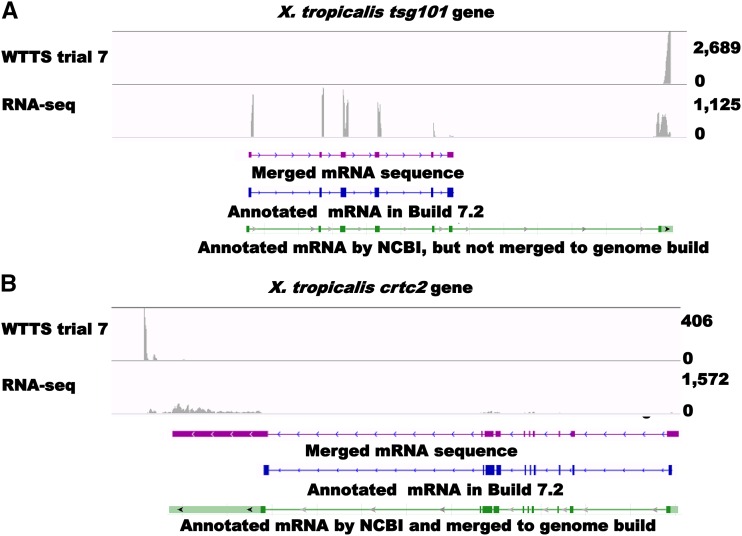
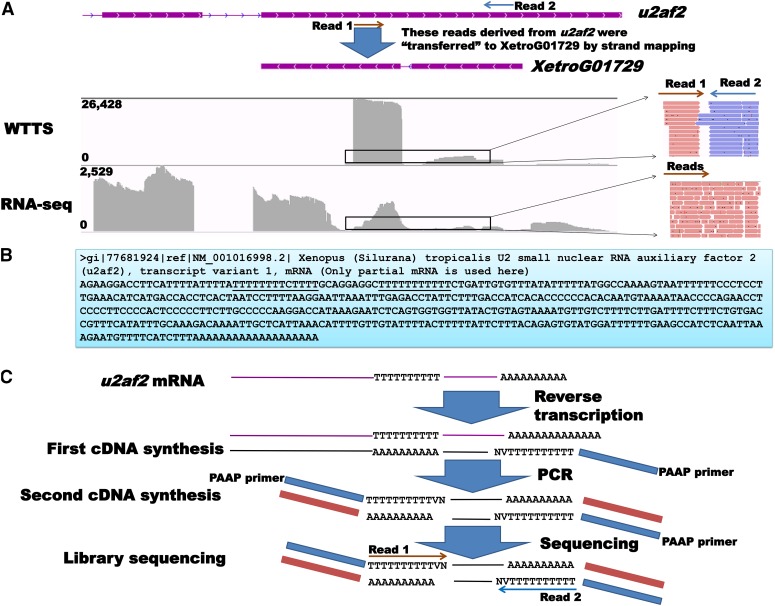
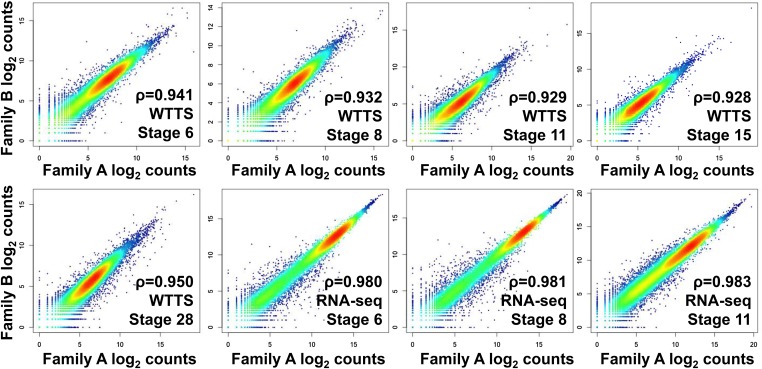
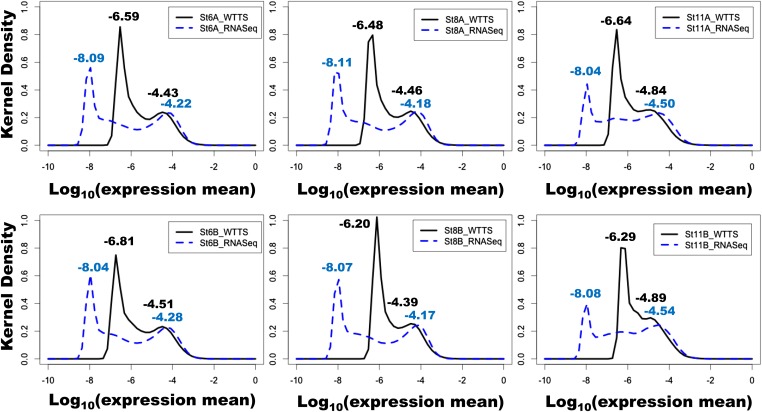
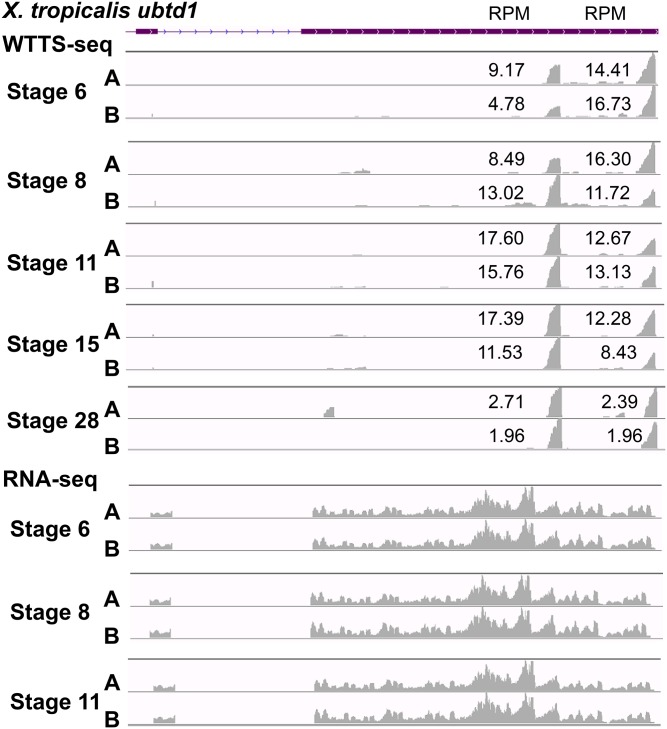
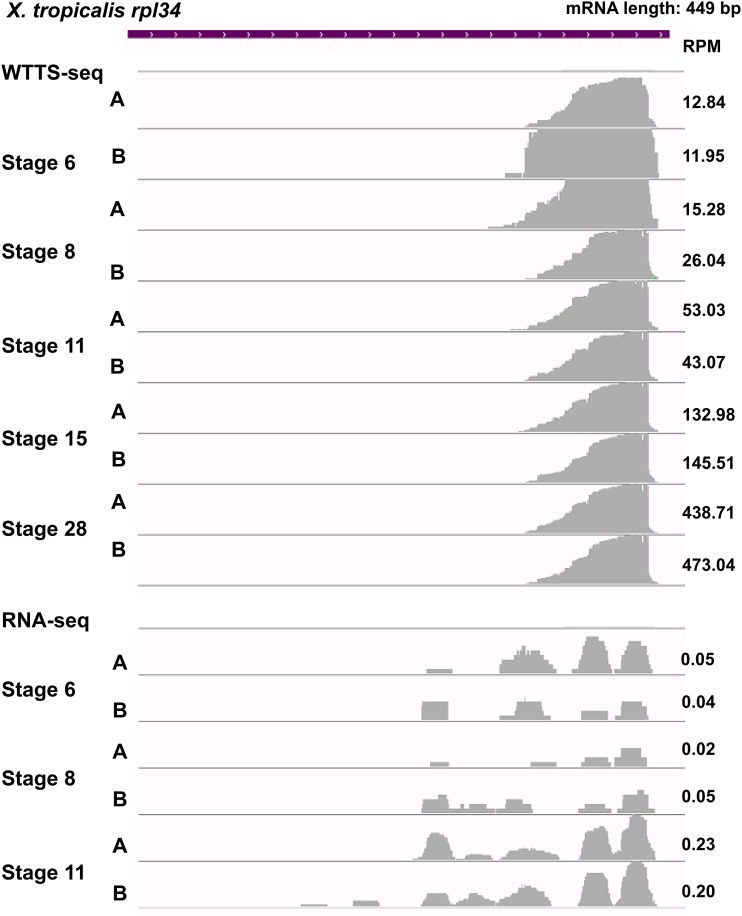
Construction of next-generation sequencing (NGS) libraries involves RNA manipulation, which often creates noisy, biased, and artifactual data that contribute to errors in transcriptome analysis. In this study, a total of 19 whole transcriptome termini site sequencing (WTTS-seq) and seven RNA sequencing (RNA-seq) libraries were prepared from Xenopus tropicalis adult and embryo samples to determine the most effective library preparation method to maximize transcriptomics investigation. We strongly suggest that appropriate primers/adaptors are designed to inhibit amplification detours and that PCR overamplification is minimized to maximize transcriptome coverage. Furthermore, genome annotation must be improved so that missing data can be recovered. In addition, a complete understanding of sequencing platforms is critical to limit the formation of false-positive results. Technically, the WTTS-seq method enriches both poly(A)+ RNA and complementary DNA, adds 5'- and 3'-adaptors in one step, pursues strand sequencing and mapping, and profiles both gene expression and alternative polyadenylation (APA). Although RNA-seq is cost prohibitive, tends to produce false-positive results, and fails to detect APA diversity and dynamics, its combination with WTTS-seq is necessary to validate transcriptome-wide APA.
新一代测序(NGS)文库的构建涉及RNA操作,这通常会产生有噪声、有偏差和人为的数据,从而导致转录组分析中的错误。在本研究中,从热带爪蟾的成体和胚胎样本中总共制备了19个全转录组末端位点测序(WTTS-seq)文库和7个RNA测序(RNA-seq)文库,以确定最有效的文库制备方法,从而最大限度地开展转录组学研究。我们强烈建议设计合适的引物/接头以抑制扩增弯路,并尽量减少PCR过度扩增,以最大限度地提高转录组覆盖率。此外,必须改进基因组注释,以便能够恢复缺失的数据。此外,全面了解测序平台对于限制假阳性结果的形成至关重要。从技术上讲,WTTS-seq方法富集了多聚腺苷酸(poly(A)+)RNA和互补DNA,一步添加5'和3'接头,进行链测序和定位,并分析基因表达和可变聚腺苷酸化(APA)。虽然RNA-seq成本高昂,容易产生假阳性结果,并且无法检测APA的多样性和动态变化,但将其与WTTS-seq结合对于验证全转录组APA是必要的。