Marjoram Paul, Wall Jeff D
Department of Preventive Medicine, University of Southern California, Los Angeles, CA 90089-9011, USA.
BMC Genet. 2006 Mar 15;7:16. doi: 10.1186/1471-2156-7-16.
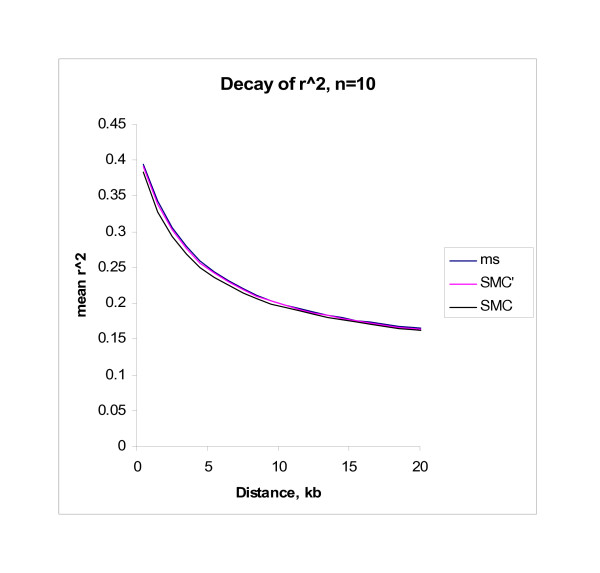
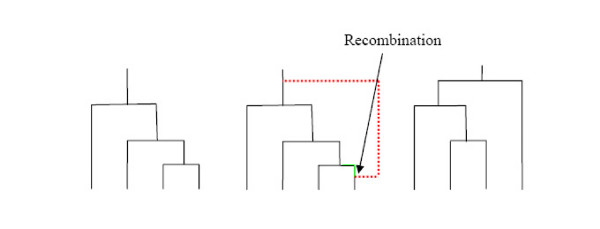
The amount of genome-wide molecular data is increasing rapidly, as is interest in developing methods appropriate for such data. There is a consequent increasing need for methods that are able to efficiently simulate such data. In this paper we implement the sequentially Markovian coalescent algorithm described by McVean and Cardin and present a further modification to that algorithm which slightly improves the closeness of the approximation to the full coalescent model. The algorithm ignores a class of recombination events known to affect the behavior of the genealogy of the sample, but which do not appear to affect the behavior of generated samples to any substantial degree.
We show that our software is able to simulate large chromosomal regions, such as those appropriate in a consideration of genome-wide data, in a way that is several orders of magnitude faster than existing coalescent algorithms.
This algorithm provides a useful resource for those needing to simulate large quantities of data for chromosomal-length regions using an approach that is much more efficient than traditional coalescent models.
全基因组分子数据量正在迅速增加,人们对开发适用于此类数据的方法的兴趣也在增加。因此,对能够有效模拟此类数据的方法的需求也日益增长。在本文中,我们实现了由麦克维恩(McVean)和卡丹(Cardin)描述的顺序马尔可夫合并算法,并对该算法进行了进一步修改,这略微提高了其对完整合并模型的近似程度。该算法忽略了一类已知会影响样本系谱行为的重组事件,但这些事件似乎在很大程度上并不影响生成样本的行为。
我们表明,我们的软件能够以比现有合并算法快几个数量级的方式模拟大型染色体区域,例如在考虑全基因组数据时适用的区域。
该算法为那些需要使用比传统合并模型高效得多的方法来模拟染色体长度区域的大量数据的人提供了有用的资源。