Aanensen David M, Feil Edward J, Holden Matthew T G, Dordel Janina, Yeats Corin A, Fedosejev Artemij, Goater Richard, Castillo-Ramírez Santiago, Corander Jukka, Colijn Caroline, Chlebowicz Monika A, Schouls Leo, Heck Max, Pluister Gerlinde, Ruimy Raymond, Kahlmeter Gunnar, Åhman Jenny, Matuschek Erika, Friedrich Alexander W, Parkhill Julian, Bentley Stephen D, Spratt Brian G, Grundmann Hajo
Department of Infectious Disease Epidemiology, School of Public Health, Imperial College London, London, United Kingdom The Centre for Genomic Pathogen Surveillance, Wellcome Genome Campus, Hinxton, Cambridge, United Kingdom.
The Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, United Kingdom.
mBio. 2016 May 5;7(3):e00444-16. doi: 10.1128/mBio.00444-16.
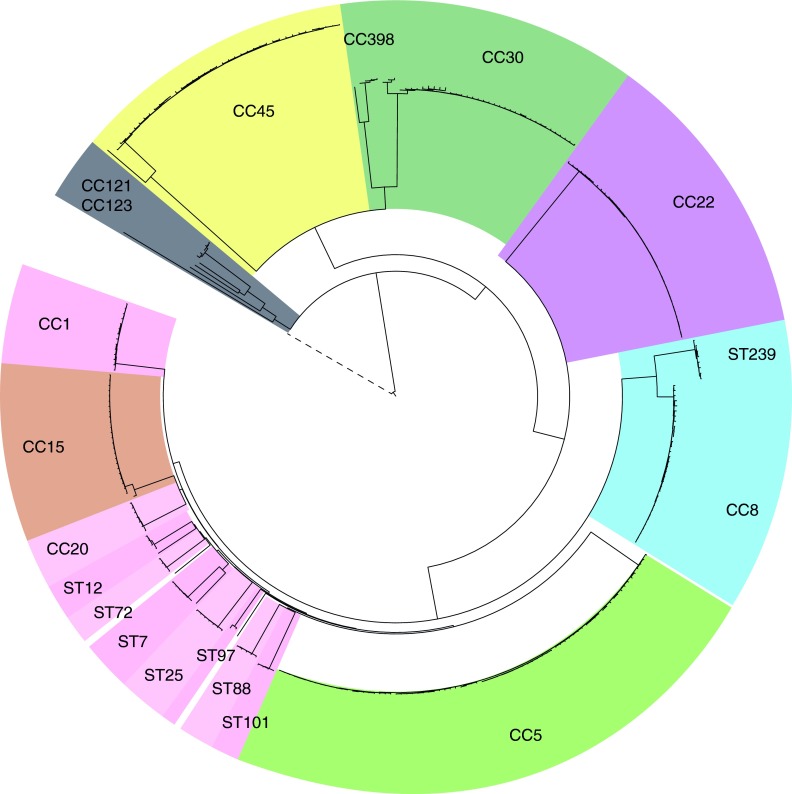
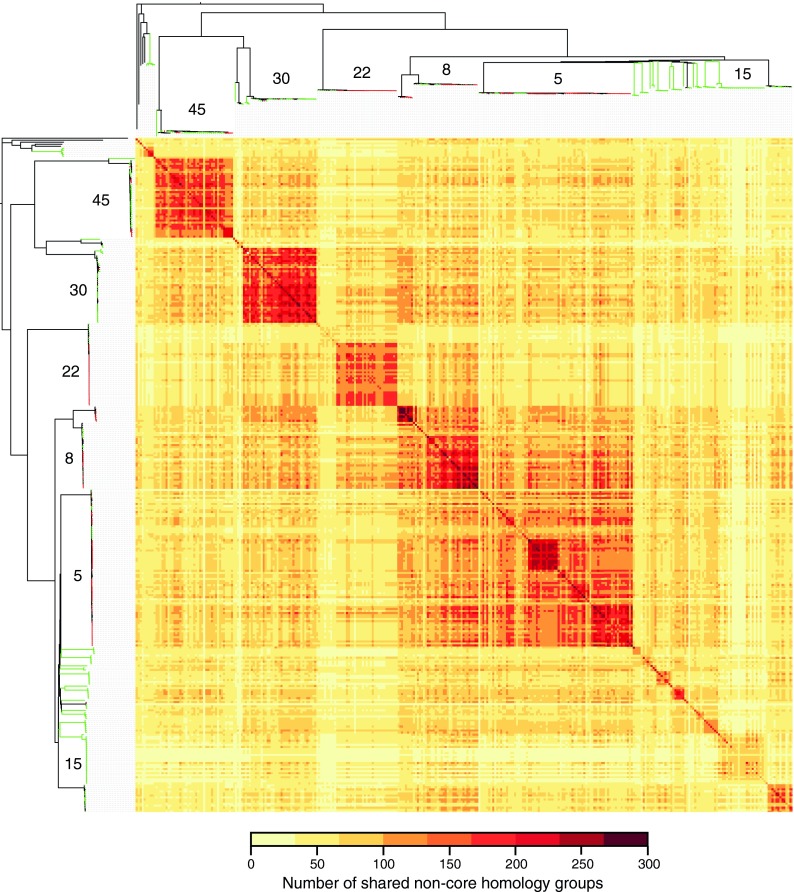
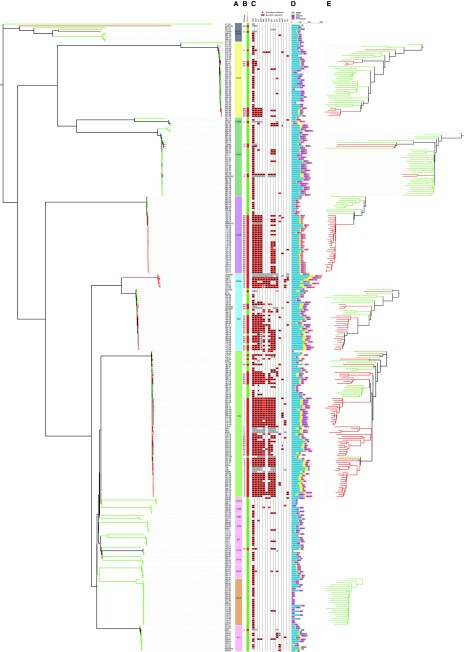
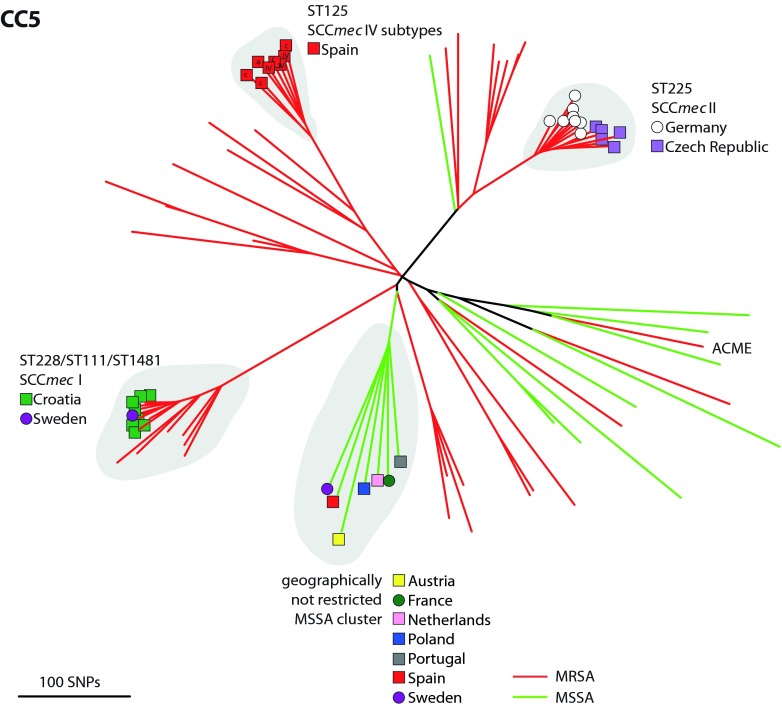
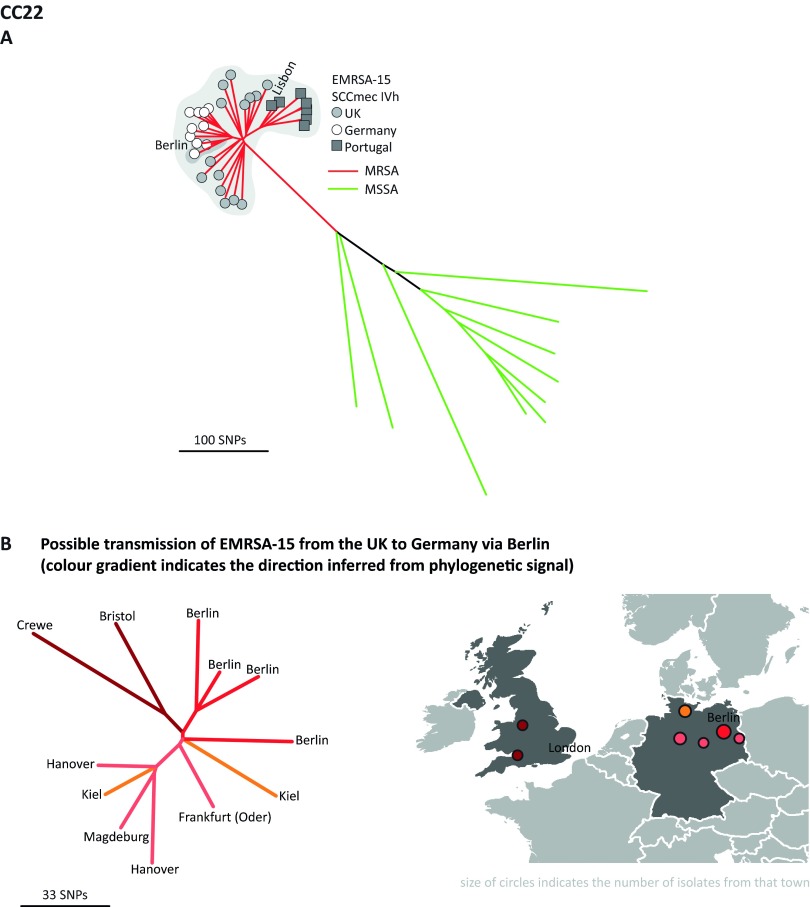
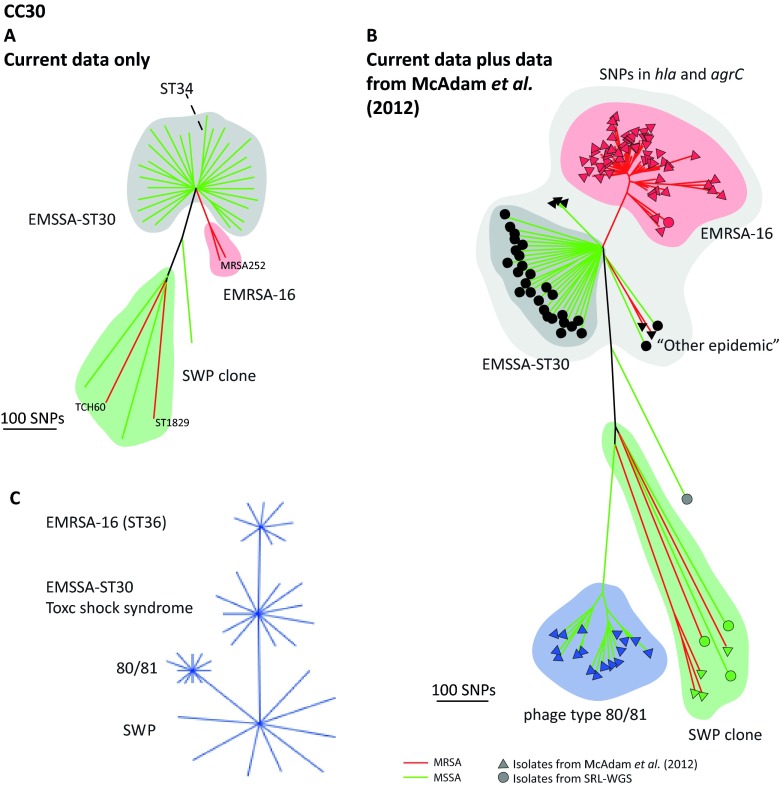
The implementation of routine whole-genome sequencing (WGS) promises to transform our ability to monitor the emergence and spread of bacterial pathogens. Here we combined WGS data from 308 invasive Staphylococcus aureus isolates corresponding to a pan-European population snapshot, with epidemiological and resistance data. Geospatial visualization of the data is made possible by a generic software tool designed for public health purposes that is available at the project URL (http://www.microreact.org/project/EkUvg9uY?tt=rc). Our analysis demonstrates that high-risk clones can be identified on the basis of population level properties such as clonal relatedness, abundance, and spatial structuring and by inferring virulence and resistance properties on the basis of gene content. We also show that in silico predictions of antibiotic resistance profiles are at least as reliable as phenotypic testing. We argue that this work provides a comprehensive road map illustrating the three vital components for future molecular epidemiological surveillance: (i) large-scale structured surveys, (ii) WGS, and (iii) community-oriented database infrastructure and analysis tools.
The spread of antibiotic-resistant bacteria is a public health emergency of global concern, threatening medical intervention at every level of health care delivery. Several recent studies have demonstrated the promise of routine whole-genome sequencing (WGS) of bacterial pathogens for epidemiological surveillance, outbreak detection, and infection control. However, as this technology becomes more widely adopted, the key challenges of generating representative national and international data sets and the development of bioinformatic tools to manage and interpret the data become increasingly pertinent. This study provides a road map for the integration of WGS data into routine pathogen surveillance. We emphasize the importance of large-scale routine surveys to provide the population context for more targeted or localized investigation and the development of open-access bioinformatic tools to provide the means to combine and compare independently generated data with publicly available data sets.
实施常规全基因组测序(WGS)有望改变我们监测细菌病原体出现和传播的能力。在此,我们将来自308株侵袭性金黄色葡萄球菌分离株的WGS数据(对应泛欧洲人群快照)与流行病学和耐药性数据相结合。通过一个为公共卫生目的设计的通用软件工具,可以对数据进行地理空间可视化,该工具可在项目网址(http://www.microreact.org/project/EkUvg9uY?tt=rc)获取。我们的分析表明,可以根据群体水平特性(如克隆相关性、丰度和空间结构)以及基于基因内容推断毒力和耐药性特性来识别高风险克隆。我们还表明,抗生素耐药谱的计算机预测至少与表型检测一样可靠。我们认为这项工作提供了一个全面的路线图,阐明了未来分子流行病学监测的三个关键组成部分:(i)大规模结构化调查,(ii)WGS,以及(iii)面向社区的数据库基础设施和分析工具。
抗生素耐药细菌的传播是一个全球关注的公共卫生紧急事件,威胁着各级医疗保健中的医学干预。最近的几项研究已经证明了细菌病原体常规全基因组测序(WGS)在流行病学监测、疫情检测和感染控制方面的前景。然而,随着这项技术的更广泛应用,生成具有代表性的国家和国际数据集以及开发用于管理和解释数据的生物信息学工具等关键挑战变得越来越相关。本研究为将WGS数据整合到常规病原体监测中提供了一个路线图。我们强调大规模常规调查对于为更有针对性或局部性调查提供群体背景的重要性,以及开发开放获取的生物信息学工具以提供将独立生成的数据与公开可用数据集进行合并和比较的手段的重要性。