Pancaldi Vera, Carrillo-de-Santa-Pau Enrique, Javierre Biola Maria, Juan David, Fraser Peter, Spivakov Mikhail, Valencia Alfonso, Rico Daniel
Structural Biology and BioComputing Programme, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
Nuclear Dynamics Programme, The Babraham Institute, Cambridge, UK.
Genome Biol. 2016 Jul 8;17(1):152. doi: 10.1186/s13059-016-1003-3.
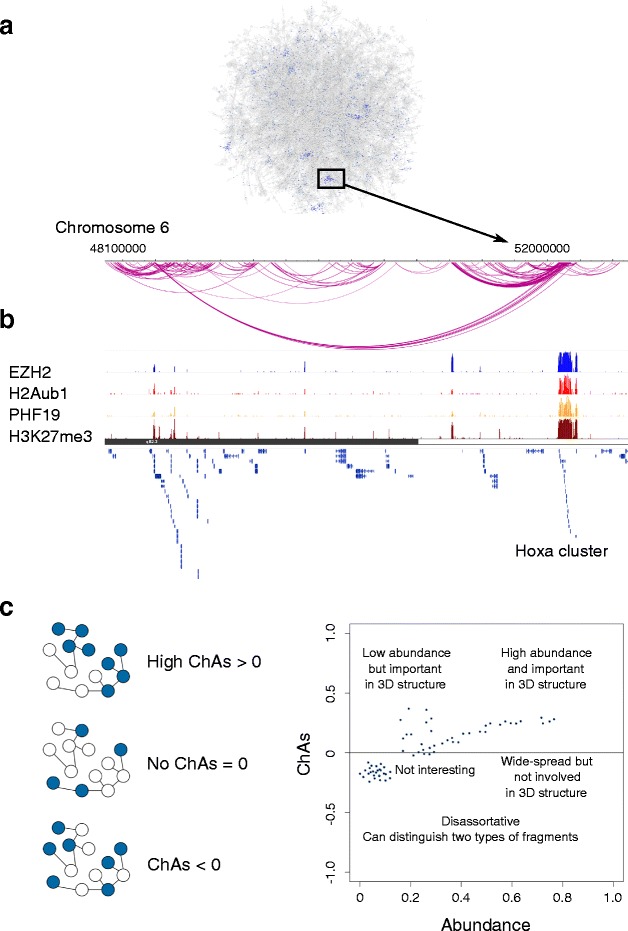
Network analysis is a powerful way of modeling chromatin interactions. Assortativity is a network property used in social sciences to identify factors affecting how people establish social ties. We propose a new approach, using chromatin assortativity, to integrate the epigenomic landscape of a specific cell type with its chromatin interaction network and thus investigate which proteins or chromatin marks mediate genomic contacts.
We use high-resolution promoter capture Hi-C and Hi-Cap data as well as ChIA-PET data from mouse embryonic stem cells to investigate promoter-centered chromatin interaction networks and calculate the presence of specific epigenomic features in the chromatin fragments constituting the nodes of the network. We estimate the association of these features with the topology of four chromatin interaction networks and identify features localized in connected areas of the network. Polycomb group proteins and associated histone marks are the features with the highest chromatin assortativity in promoter-centered networks. We then ask which features distinguish contacts amongst promoters from contacts between promoters and other genomic elements. We observe higher chromatin assortativity of the actively elongating form of RNA polymerase 2 (RNAPII) compared with inactive forms only in interactions between promoters and other elements.
Contacts among promoters and between promoters and other elements have different characteristic epigenomic features. We identify a possible role for the elongating form of RNAPII in mediating interactions among promoters, enhancers, and transcribed gene bodies. Our approach facilitates the study of multiple genome-wide epigenomic profiles, considering network topology and allowing the comparison of chromatin interaction networks.
网络分析是一种对染色质相互作用进行建模的强大方法。 assortativity是社会科学中用于识别影响人们建立社会关系因素的一种网络属性。我们提出了一种新方法,利用染色质assortativity,将特定细胞类型的表观基因组景观与其染色质相互作用网络整合起来,从而研究哪些蛋白质或染色质标记介导基因组接触。
我们使用来自小鼠胚胎干细胞的高分辨率启动子捕获Hi-C和Hi-Cap数据以及ChIA-PET数据,来研究以启动子为中心的染色质相互作用网络,并计算构成网络节点的染色质片段中特定表观基因组特征的存在情况。我们估计这些特征与四个染色质相互作用网络拓扑结构的关联,并识别位于网络连接区域的特征。多梳蛋白组蛋白和相关的组蛋白标记是以启动子为中心的网络中染色质assortativity最高的特征。然后,我们询问哪些特征区分启动子之间的接触与启动子与其他基因组元件之间的接触。我们观察到,仅在启动子与其他元件之间的相互作用中,与非活性形式相比,RNA聚合酶2(RNAPII)的活性延伸形式具有更高的染色质assortativity。
启动子之间以及启动子与其他元件之间的接触具有不同的特征表观基因组特征。我们确定了RNAPII延伸形式在介导启动子、增强子和转录基因体之间相互作用中的可能作用。我们的方法有助于研究多个全基因组表观基因组图谱,考虑网络拓扑结构并允许比较染色质相互作用网络。