Key Laboratory of Brain Functional Genomics of STCSM, Institute of Cognitive Neuroscience, East China Normal University, Shanghai 200062, China.
Department of Pediatrics, Division of Experimental Hematology and Cancer Biology, Cincinnati Children's Hospital Medical Center, University of Cincinnati, Cincinnati, Ohio 45229, USA.
Nat Commun. 2016 Jul 15;7:12185. doi: 10.1038/ncomms12185.
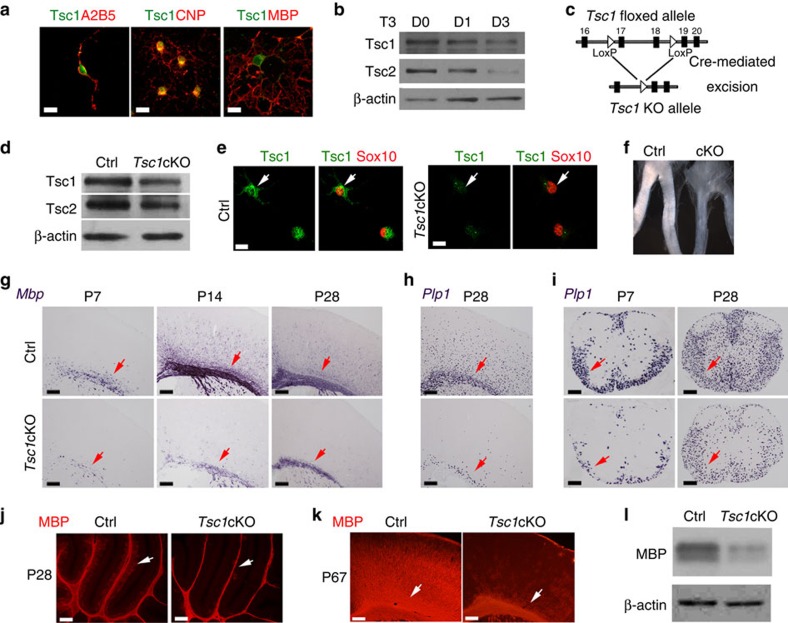
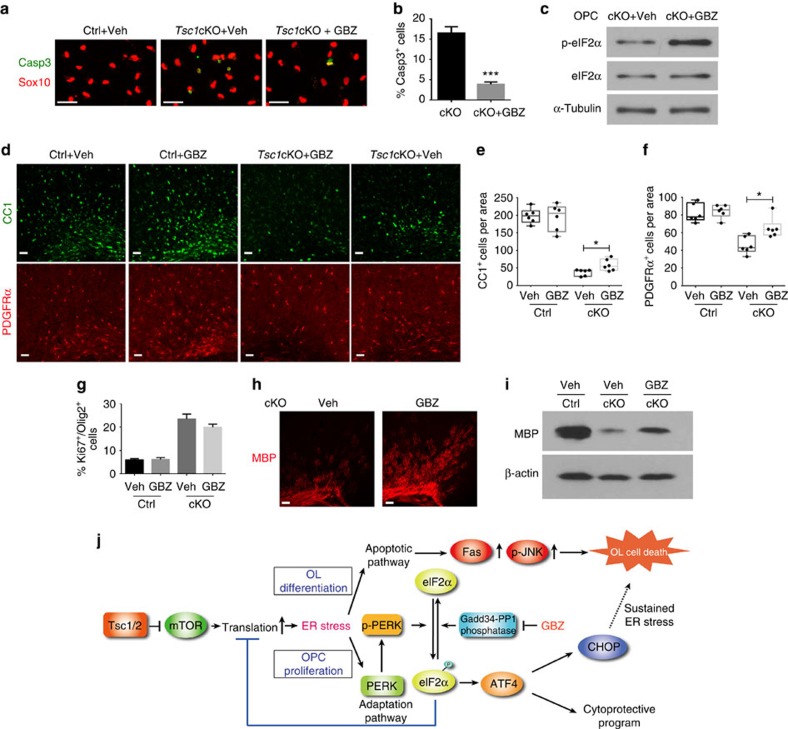
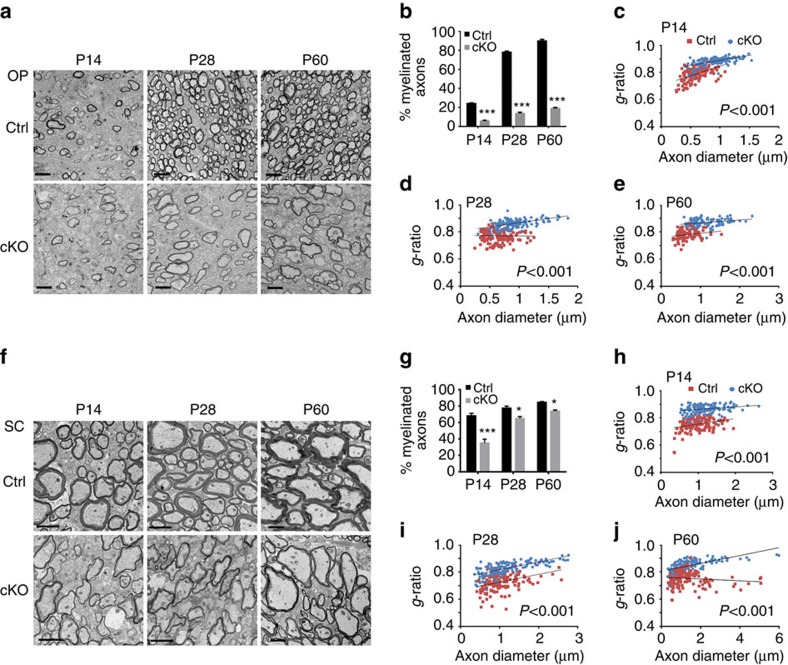
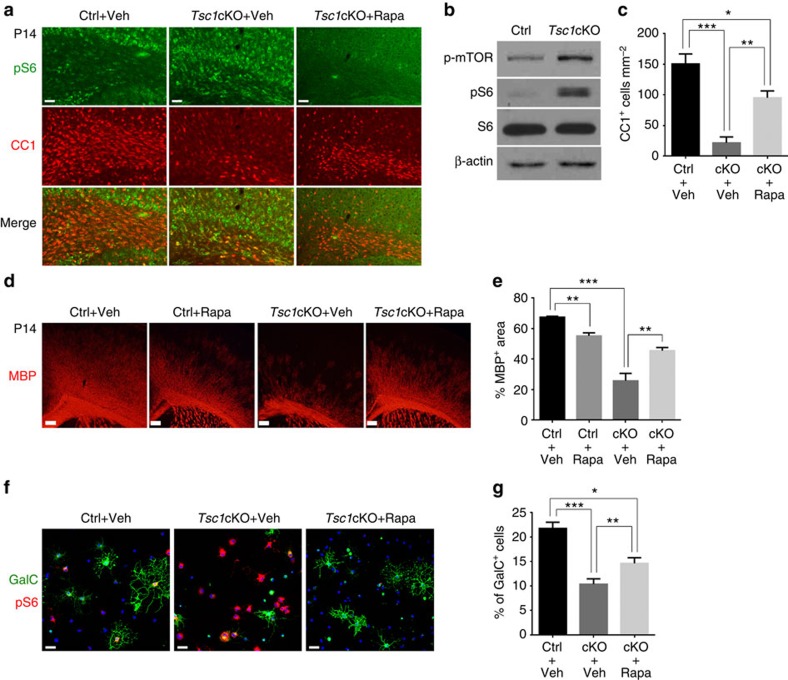
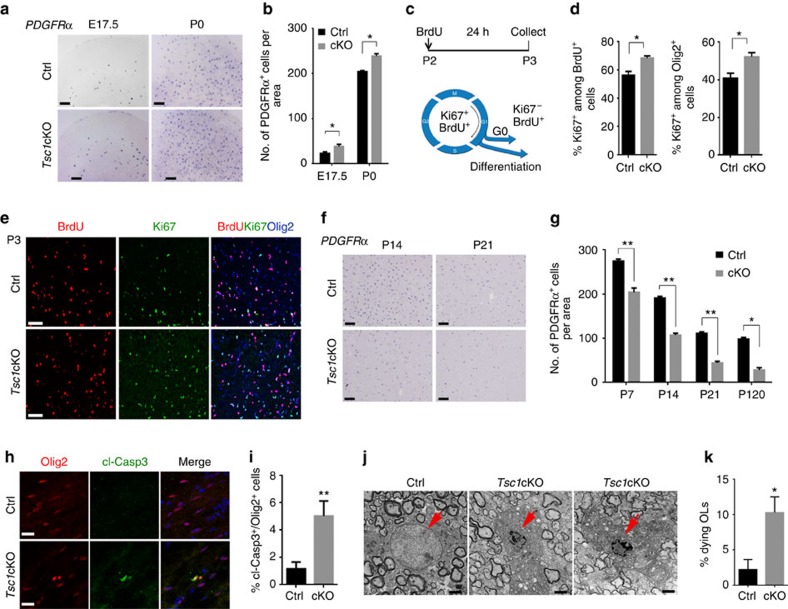
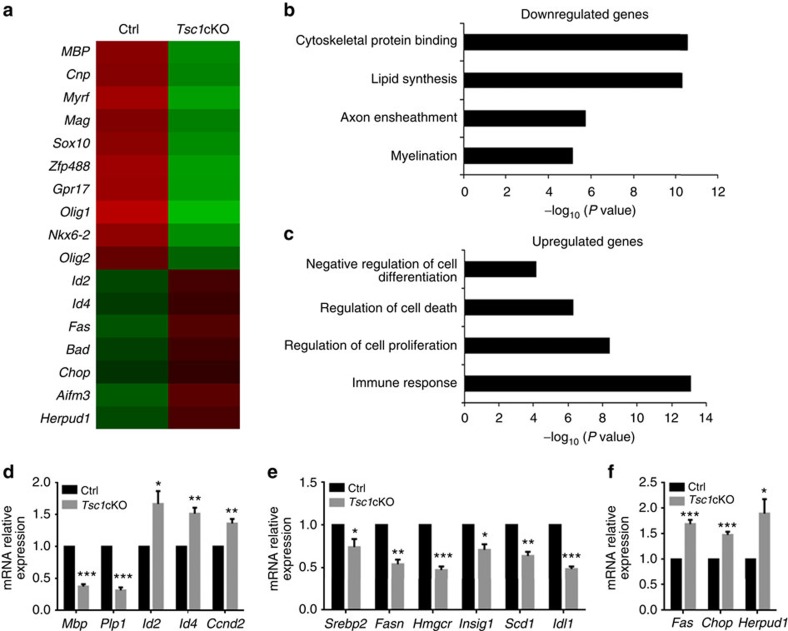
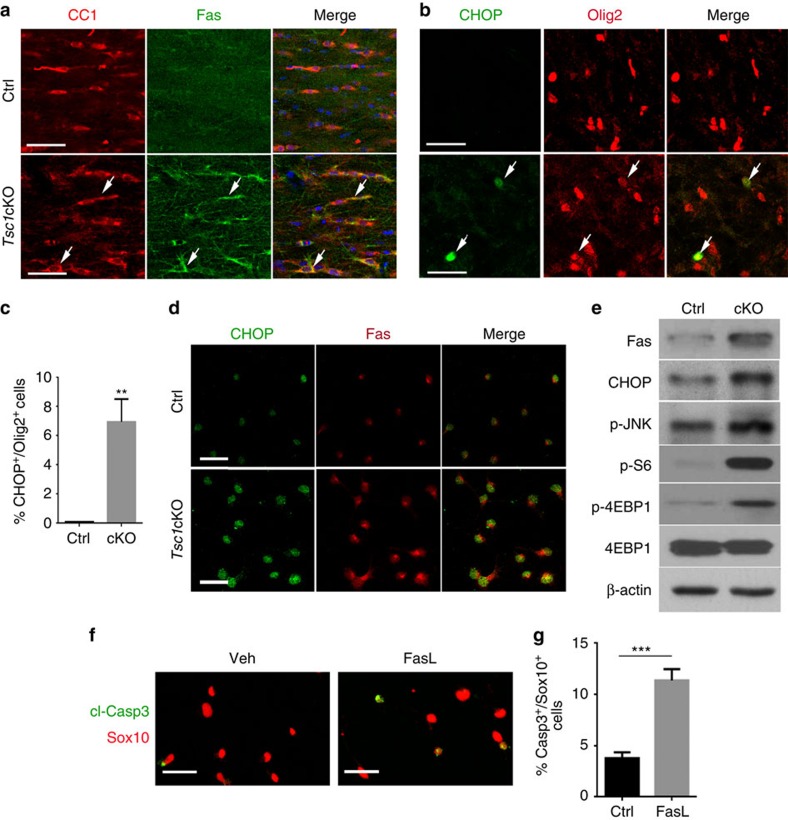
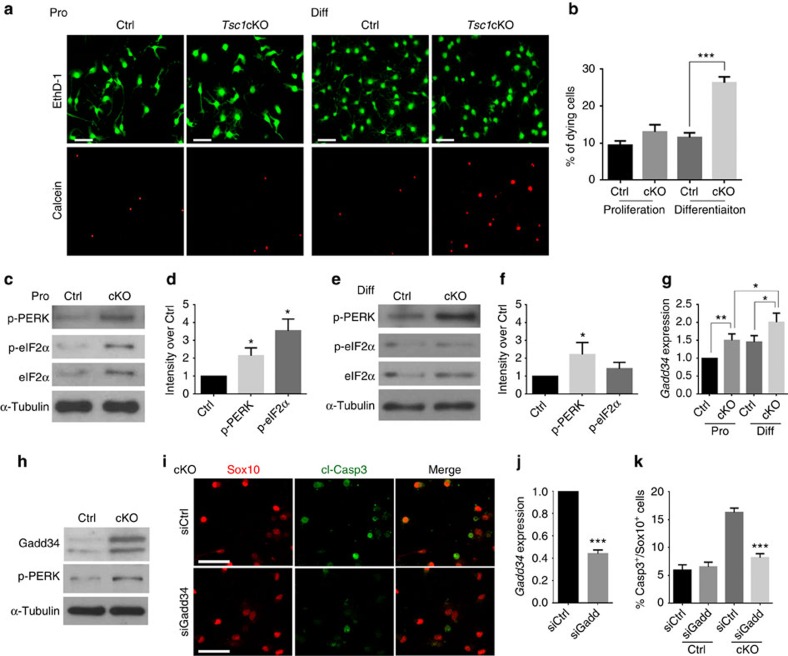
Tuberous sclerosis complex-1 or 2 (TSC1/2) mutations cause white matter abnormalities, including myelin deficits in the CNS; however, underlying mechanisms are not fully understood. TSC1/2 negatively regulate the function of mTOR, which is required for oligodendrocyte differentiation. Here we report that, unexpectedly, constitutive activation of mTOR signalling by Tsc1 deletion in the oligodendrocyte lineage results in severe myelination defects and oligodendrocyte cell death in mice, despite an initial increase of oligodendrocyte precursors during early development. Expression profiling analysis reveals that Tsc1 ablation induces prominent endoplasmic reticulum (ER) stress responses by activating a PERK-eIF2α signalling axis and Fas-JNK apoptotic pathways. Enhancement of the phospho-eIF2α adaptation pathway by inhibition of Gadd34-PP1 phosphatase with guanabenz protects oligodendrocytes and partially rescues myelination defects in Tsc1 mutants. Thus, TSC1-mTOR signalling acts as an important checkpoint for maintaining oligodendrocyte homoeostasis, pointing to a previously uncharacterized ER stress mechanism that contributes to hypomyelination in tuberous sclerosis.
结节性硬化症复合物-1 或 2(TSC1/2)突变导致白质异常,包括中枢神经系统髓鞘缺陷;然而,其潜在机制尚不完全清楚。TSC1/2 负向调节 mTOR 的功能,mTOR 是少突胶质细胞分化所必需的。在这里,我们报告说,出乎意料的是,少突胶质细胞谱系中 Tsc1 的组成性激活导致 mTOR 信号的激活导致严重的髓鞘缺陷和少突胶质细胞死亡,尽管在早期发育过程中少突胶质前体细胞最初增加。表达谱分析表明,Tsc1 缺失通过激活 PERK-eIF2α 信号轴和 Fas-JNK 凋亡途径诱导明显的内质网(ER)应激反应。用胍那苄抑制 Gadd34-PP1 磷酸酶来增强磷酸化 eIF2α 的适应途径,可以保护少突胶质细胞,并部分挽救 Tsc1 突变体中的髓鞘缺陷。因此,TSC1-mTOR 信号作为维持少突胶质细胞稳态的重要检查点,指出了一种以前未被描述的 ER 应激机制,该机制导致结节性硬化症中的髓鞘减少。